木聚糖基碳基固体酸及由其催化合成苯并呫吨类化合物的方法与流程
本发明属于有机催化合成技术领域,具体涉及一种木聚糖基碳基固体酸及由其催化合成苯并呫吨类化合物的方法。
背景技术:
含氧杂环化合物广泛存在于各种天然产物和药物活性分子中,同时也是重要的有机合成中间体。含氧杂环的合成与转化对医学开发和天然产物合成等领域具有重要意义。呫吨作为一类重要的含氧杂环化合物,早期主要被用作化学荧光燃料应用于激光技术和生物分子荧光标记等领域。随着研究的深入发展,研究者发现呫吨,特别是苯并呫吨类化合物具有丰富的生物活性和药用价值,如在医学上具有抗病毒、抗氧化、抗肿瘤、消炎镇痛等作用。苯并呫吨类化合物通常以醛类化合物、2-萘酚和1,3-环己二酮类化合物在催化剂的作用下通过缩合反应制备。目前报道的催化剂种类很多,如InCl3、硝酸铈铵、对甲基苯磺酸、负载型离子液体、脯氨酸三氟甲磺酸酯、NaHSO4·SiO2等。但是,这些反应体系或多或少存在着不可避免的缺点,如反应时间长,产率低,使用有毒的有机溶剂,催化剂污染环境及重复使用难以实现等。
随着人们对环保和绿色化工过程的关注度越来越高,人们选择更有前景的固体酸催化剂代替液体无机酸。碳基固体酸催化剂是一种新型的固体酸催化剂,可以解决目前工业上采用液体无机酸(如硫酸和盐酸等)作为酸催化剂时存在的产物分离困难、腐蚀设备、污染环境等问题。近年来,因碳基固体酸具有很高的酸密度和热稳定性,并且其表面具有良好的疏水性,其酸活性位不会被水溶剂或反应生成的水侵蚀而流失,化学稳定性较好而作为一类新型的固体酸催化剂引起了越来越多科研工作者的重视。
生物质资源因其丰富的含碳量被认为是一种制备碳基固体酸催化剂良好的原材料。木聚糖是许多植物如玉米芯、棉籽壳、木屑、甘蔗渣和桦木等细胞壁半纤维素的主要成分,是一种取之不尽、用之不竭的可再生性植物资源。木聚糖具有特殊的化学结构,如分枝、无定型组成的几种不同类型的单糖(杂多糖)和不同类型的官能团(如乙酰基、羟基、羧基、甲氧基)等。分子结构的复杂性和多样性决定了木聚糖具有独特的活性,被广泛应用于食品添加剂、造纸以及生物医学等领域。
技术实现要素:
为了解决现有催化剂的缺点和不足之处,本发明的首要目的在于提供一种木聚糖基碳基固体酸催化剂的制备方法。
本发明的另一目的在于提供一种通过上述方法制备得到的木聚糖基碳基固体酸催化剂。
本发明的再一目的在于提供一种采用上述木聚糖基碳基固体酸催化剂催化合成苯并呫吨类化合物的方法。
本发明目的通过以下技术方案实现:
一种木聚糖基碳基固体酸催化剂的制备方法,包括如下制备步骤:
(1)将木聚糖类半纤维素在氮气氛围下,从室温加热到200~400℃,保温反应60~180min,冷却至室温,得到黑色粉末状的木聚糖类半纤维素不完全碳化产物;
(2)将步骤(1)所得木聚糖类半纤维素不完全碳化产物与浓硫酸加入到反应器中,氮气氛围下升温至100~250℃反应12~18h;
(3)将步骤(2)所得反应混合物在搅拌条件下滴加到冰水混合物中,过滤,固体物质经洗涤、干燥,得到所述木聚糖基碳基固体酸催化剂。
优选地,步骤(1)中所述反应温度为350℃,反应时间为90min。
优选地,步骤(2)中所述木聚糖类半纤维素不完全碳化产物与浓硫酸的质量体积比为1:30g/mL。
优选地,步骤(2)中所述反应温度为150℃,反应时间为15h。
优选地,步骤(3)中所述的洗涤是指用>80℃的热水洗涤至洗脱液呈中性;所述的干燥是指在80℃下真空干燥。
一种木聚糖基碳基固体酸催化剂,通过上述方法制备得到。
优选地,所述木聚糖基碳基固体酸催化剂的磺酸含量为1.08mmol/g。
一种采用上述木聚糖基碳基固体酸催化剂催化合成苯并呫吨类化合物的方法,包括如下步骤:
将木聚糖基碳基固体酸催化剂、醛类化合物、2-萘酚和1,3-环己二酮类化合物加入到反应器中,70~110℃油浴下催化反应1~3h;过滤除去催化剂,滤液减压蒸馏除去溶剂,经柱层析或重结晶提纯,得到所述苯并呫吨类化合物。
优选地,所述木聚糖基碳基固体酸催化剂的用量为4.32mol%。
优选地,所述催化反应温度为90℃,反应时间为2h。
上述合成反应原理如下式所示:
相对于现有技术,本发明具有如下优点及有益效果:
(1)本发明采用廉价、无毒、可再生、可生物降解与生物相容性良好的木聚糖类半纤维素为原料制备木聚糖基碳基固体酸催化剂,有利于环境保护和减少含有过渡金属离子的催化剂的使用;
(2)本发明木聚糖基碳基固体酸催化剂的制备方法操作简单,成本低,反应条件易于控制;
(3)本发明制备的木聚糖基碳基固体酸作为催化剂,具有环境友好性、生物兼容性、高催化活性及可循环使用性等优点。
附图说明
图1为本发明实施例1~9所得12-苯基-9,9-二甲基-8,9,10,12-苯并[a]吨-11-酮的核磁氢谱图。
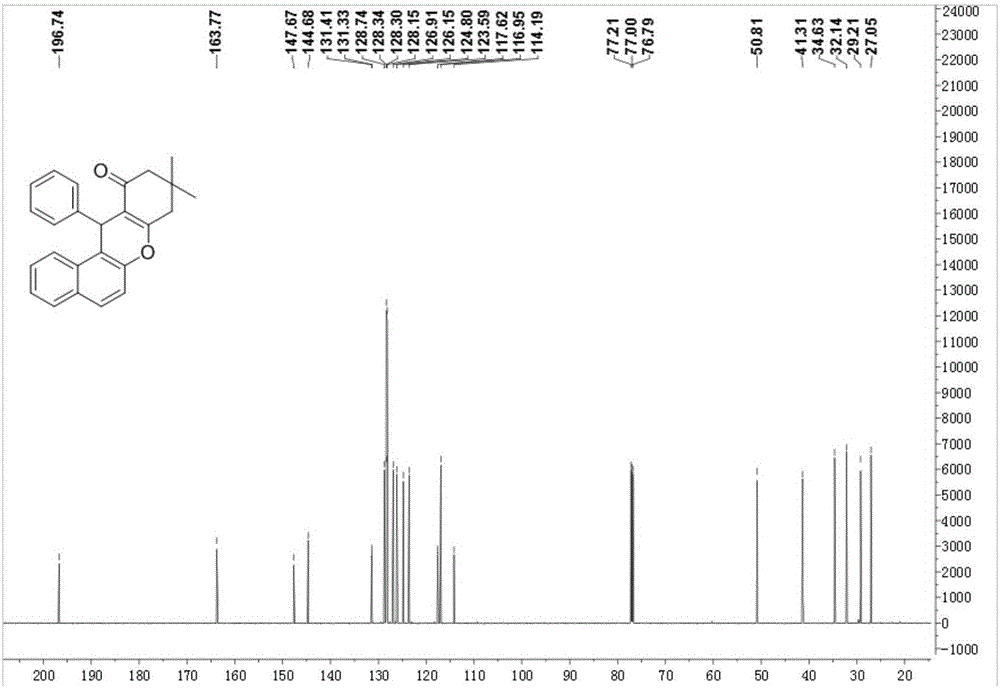
图2为本发明实施例1~9所得12-苯基-9,9-二甲基-8,9,10,12-苯并[a]吨-11-酮的核磁碳谱图。
图3为本发明新制备的木聚糖基碳基固体酸催化剂(a)与重复使用5次后的催化剂(b)的红外光谱图。
图4为本发明新制备的木聚糖基碳基固体酸催化剂(a)与重复使用5次后的催化剂(b)的XRD谱图。
具体实施方式
下面结合实施例及附图对本发明作进一步详细的描述,但本发明的实施方式不限于此。
实施例1
(1)准确称取3g木聚糖类半纤维素于坩埚中置于管式炉中,在氮气氛围下,从室温加热到200℃保温反应180min,待反应结束后,冷却至室温,得到黑色粉末状的木聚糖类半纤维素不完全碳化产物;
(2)将步骤(1)得到的产物与浓硫酸按照1:30(g:mL)的比例混合后置于带有聚四氟乙烯内衬的反应釜中,在氮气氛围下,将该反应体系从室温加热至250℃反应12h;
(3)将步骤(2)所得反应混合物在不断搅拌的条件下滴加到1000mL冰水混合物中,滴加完毕后,待溶液冷却至室温,过滤,用热水(>80℃)充分洗涤固体物质至洗脱液呈中性,然后在80℃下真空干燥,得到所述木聚糖基碳基固体酸催化剂。
实施例2
(1)准确称取3g木聚糖类半纤维素于坩埚中置于管式炉中,在氮气氛围下,从室温加热到350℃保温反应90min,待反应结束后,冷却至室温,得到黑色粉末状的木聚糖类半纤维素不完全碳化产物;
(2)将步骤(1)得到的产物与浓硫酸按照1:30(g:mL)的比例混合后置于带有聚四氟乙烯内衬的反应釜中,在氮气氛围下,将该反应体系从室温加热至150℃反应15h;
(3)将步骤(2)所得反应混合物在不断搅拌的条件下滴加到1000mL冰水混合物中,滴加完毕后,待溶液冷却至室温,过滤,用热水(>80℃)充分洗涤固体物质至洗脱液呈中性,然后在80℃下真空干燥,得到所述木聚糖基碳基固体酸催化剂。
实施例3
(1)准确称取3g木聚糖类半纤维素于坩埚中置于管式炉中,在氮气氛围下,从室温加热到400℃保温反应60min,待反应结束后,冷却至室温,得到黑色粉末状的木聚糖类半纤维素不完全碳化产物;
(2)将步骤(1)得到的产物与浓硫酸按照1:30(g:mL)的比例混合后置于带有聚四氟乙烯内衬的反应釜中,在氮气氛围下,将该反应体系从室温加热至100℃反应18h;
(3)将步骤(2)所得反应混合物在不断搅拌的条件下滴加到1000mL冰水混合物中,滴加完毕后,待溶液冷却至室温,过滤,用热水(>80℃)充分洗涤固体物质至洗脱液呈中性,然后在80℃下真空干燥,得到所述木聚糖基碳基固体酸催化剂。
实施例4
(1)取20mg实施例2所得木聚糖基碳基固体酸催化剂(炭化时间90min,炭化温度350℃,磺化时间15h,磺化温度150℃),磺酸含量为1.08mmol/g于反应容器中,然后相继加入苯甲醛(0.5mmol),2-萘酚(0.5mmol),5,5-二甲基环己二酮(0.6mmol);
(2)将步骤(1)体系中加入一颗磁子,于90℃下反应2h;
(3)将产物冷却至室温,倾入适量乙酸乙酯,沉淀出催化剂,过滤,用乙酸乙酯洗涤数次,将得到的催化剂在真空80℃下干燥24h后直接用于下次反应;
(4)将步骤(3)得到的有机层减压蒸馏得到粗产物,粗产物经柱层析分离或重结晶得到纯净的12-苯基-9,9-二甲基-8,9,10,12-苯并[a]吨-11-酮,产率为74%。
实施例5
(1)取30mg实施例2所得木聚糖基碳基固体酸催化剂(炭化时间90min,炭化温度350℃,磺化时间15h,磺化温度150℃),磺酸含量为1.08mmol/g于反应容器中,然后相继加入苯甲醛(0.5mmol),2-萘酚(0.5mmol),5,5-二甲基环己二酮(0.6mmol);
(2)将步骤(1)体系中加入一颗磁子,于90℃下反应2h;
(3)将产物冷却至室温,倾入适量乙酸乙酯,沉淀出催化剂,过滤,用乙酸乙酯洗涤数次,将得到的催化剂在真空80℃下干燥24h后直接用于下次反应;
(4)将步骤(3)得到的有机层减压蒸馏得到粗产物,粗产物经柱层析分离或重结晶得到纯净的12-苯基-9,9-二甲基-8,9,10,12-苯并[a]吨-11-酮,产率为85%。
实施例6
(1)取40mg实施例2所得木聚糖基碳基固体酸催化剂(炭化时间90min,炭化温度350℃,磺化时间15h,磺化温度150℃),磺酸含量为1.08mmol/g于反应容器中,然后相继加入苯甲醛(0.5mmol),2-萘酚(0.5mmol),5,5-二甲基环己二酮(0.6mmol);
(2)将步骤(1)体系中加入一颗磁子,于90℃下反应2h;
(3)将产物冷却至室温,倾入适量乙酸乙酯,沉淀出催化剂,过滤,用乙酸乙酯洗涤数次,将得到的催化剂在真空80℃下干燥24h后直接用于下次反应;
(4)将步骤(3)得到的有机层减压蒸馏得到粗产物,粗产物经柱层析分离或重结晶得到纯净的12-苯基-9,9-二甲基-8,9,10,12-苯并[a]吨-11-酮,产率为88%。
实施例7
(1)取50mg实施例2所得木聚糖基碳基固体酸催化剂(炭化时间90min,炭化温度350℃,磺化时间15h,磺化温度150℃),磺酸含量为1.08mmol/g于反应容器中,然后相继加入苯甲醛(0.5mmol),2-萘酚(0.5mmol),5,5-二甲基环己二酮(0.6mmol);
(2)将步骤(1)体系中加入一颗磁子,于90℃下反应2h;
(3)将产物冷却至室温,倾入适量乙酸乙酯,沉淀出催化剂,过滤,用乙酸乙酯洗涤数次,将得到的催化剂在真空80℃下干燥24h后直接用于下次反应;
(4)将步骤(3)得到的有机层减压蒸馏得到粗产物,粗产物经柱层析分离或重结晶得到纯净的12-苯基-9,9-二甲基-8,9,10,12-苯并[a]吨-11-酮,产率为89%。
实施例8
(1)取40mg实施例2所得木聚糖基碳基固体酸催化剂(炭化时间90min,炭化温度350℃,磺化时间15h,磺化温度150℃),磺酸含量为1.08mmol/g于反应容器中,然后相继加入苯甲醛(0.5mmol),2-萘酚(0.5mmol),5,5-二甲基环己二酮(0.6mmol);
(2)将步骤(1)体系中加入一颗磁子,于80℃下反应3h;
(3)将产物冷却至室温,倾入适量乙酸乙酯,沉淀出催化剂,过滤,用乙酸乙酯洗涤数次,将得到的催化剂在真空80℃下干燥24h后直接用于下次反应;
(4)将步骤(3)得到的有机层减压蒸馏得到粗产物,粗产物经柱层析分离或重结晶得到纯净的12-苯基-9,9-二甲基-8,9,10,12-苯并[a]吨-11-酮,产率为81%。
实施例9
(1)取40mg实施例2所得木聚糖基碳基固体酸催化剂(炭化时间90min,炭化温度350℃,磺化时间15h,磺化温度150℃),磺酸含量为1.08mmol/g于反应容器中,然后相继加入苯甲醛(0.5mmol),2-萘酚(0.5mmol),5,5-二甲基环己二酮(0.6mmol);
(2)将步骤(1)体系中加入一颗磁子,于110℃下反应1h;
(3)将产物冷却至室温,倾入适量乙酸乙酯,沉淀出催化剂,过滤,用乙酸乙酯洗涤数次,将得到的催化剂在真空80℃下干燥24h后直接用于下次反应;
(4)将步骤(3)得到的有机层减压蒸馏得到粗产物,粗产物经柱层析分离或重结晶得到纯净的12-苯基-9,9-二甲基-8,9,10,12-苯并[a]吨-11-酮,产率为85%。
实施例10
(1)取40mg实施例2所得木聚糖基碳基固体酸催化剂(炭化时间90min,炭化温度350℃,磺化时间15h,磺化温度150℃),磺酸含量为1.08mmol/g于反应容器中,然后相继加入4-硝基苯甲醛(0.5mmol),2-萘酚(0.5mmol),5,5-二甲基环己二酮(0.6mmol);
(2)将步骤(1)体系中加入一颗磁子,于90℃反应2h;
(3)将产物冷却至室温,倾入适量乙酸乙酯,沉淀出催化剂,过滤,用乙酸乙酯洗涤数次,将得到的催化剂在真空80℃下干燥24h后直接用于下次反应;
(4)将步骤(3)得到的有机层减压蒸馏得到粗产物,粗产物经柱层析分离或重结晶得到纯净的产物,产率为96%。
实施例11
(1)取40mg实施例2所得木聚糖基碳基固体酸催化剂(炭化时间90min,炭化温度350℃,磺化时间15h,磺化温度150℃),磺酸含量为1.08mmol/g于反应容器中,然后相继加入乙醛(0.5mmol),2-萘酚(0.5mmol),5,5-二甲基环己二酮(0.6mmol);
(2)将步骤(1)体系中加入一颗磁子,于90℃反应2h;
(3)将产物冷却至室温,倾入适量乙酸乙酯,沉淀出催化剂,过滤,用乙酸乙酯洗涤数次,将得到的催化剂在真空80℃下干燥24h后直接用于下次反应;
(4)将步骤(3)得到的有机层减压蒸馏得到粗产物,粗产物经柱层析分离或重结晶得到纯净的产物,产率为80%。
实施例12
(1)取40mg实施例2所得木聚糖基碳基固体酸催化剂(炭化时间90min,炭化温度350℃,磺化时间15h,磺化温度150℃),磺酸含量为1.08mmol/g于反应容器中,然后相继加入4-硝基苯甲醛(0.5mmol),2-萘酚(0.5mmol),环己二酮(0.6mmol);
(2)将步骤(1)体系中加入一颗磁子,于90℃反应2h;
(3)将产物冷却至室温,倾入适量乙酸乙酯,沉淀出催化剂,过滤,用乙酸乙酯洗涤数次,将得到的催化剂在真空80℃下干燥24h后直接用于下次反应;
(4)将步骤(3)得到的有机层减压蒸馏得到粗产物,粗产物经柱层析分离或重结晶得到纯净的产物,产率为95%。
以上实施例1~9所得12-苯基-9,9-二甲基-8,9,10,12-苯并[a]吨-11-酮的核磁氢谱图如图1所示。由图1可以看出,在12-苯基-9,9-二甲基-8,9,10,12-苯并[a]吨-11-酮的核磁氢谱图中,8.02处为苯环上面的氢,为双重峰,耦合常数J=12.0Hz,7.98-7.76为苯环上的2个H,为多重峰,7.45-7.34为苯环上的5个H,为多重峰,7.21-7.18为苯环上的2个H,为多重峰,7.09-7.06为苯环上的1个H,为多重峰,5.75为与苯环相连的C上的一个H,为单重峰,2.57为六元环中远离C=O双键的C上的两个氢,为单重峰,2.28为六元环中靠近C=O双键的C上的两个氢,为两个双重峰,耦合常数J1=18.0Hz,J2=36.0Hz,1.12和0.97处分别为六元环上两个甲基处的3个氢,为单重峰。
以上实施例1~9所得12-苯基-9,9-二甲基-8,9,10,12-苯并[a]吨-11-酮的核磁碳谱图如图2所示。由图2可以看出,在12-苯基-9,9-二甲基-8,9,10,12-苯并[a]吨-11-酮的核磁碳谱图的中196.74,163.77,147.67,144.68,131.41,131.33,128.74,128.34,128.30,128.15,126.91,126.15,124.80,123.59,117.62,116.95,114.19,50.81,41.31,34.63,32.14,26.21,27.05分别对应相应结构中C的化学位移。
图3为本发明新制备的木聚糖基碳基固体酸催化剂(Fresh CXH-SO3H)和循环使用5次后的催化剂(5th-reused CXH-SO3H)的红外谱图。从图中可以看出,两者均在1037cm-1和1177cm-1处有SO3H基团的特征峰。两者谱图看似相似,但是仔细观察发现,新鲜制备的催化剂的SO3H基团的特征峰比较宽,而使用过5次后的催化剂的SO3H基团特征峰较窄,并且与新鲜制备的催化剂有略微的偏移。这可能是催化剂在使用过程中与有机溶剂接触过所致。
图4为本发明新制备的木聚糖基碳基固体酸催化剂(Fresh CXH-SO3H)与重复使用5次后的催化剂(5th-reused CXH-SO3H)的X射线衍射图(XRD)。从XRD图中可以看出,在2θ=20-30°处的包峰对应炭的无定型结构,木聚糖基碳基固体酸催化剂在使用前后的包峰没有发生明显的变化,说明催化剂在使用过程中的碳骨架没有遭到破坏,依然保持着无定型炭的结构。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其它的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!