本发明涉及一种催化剂制备方法,具体地说涉及一种非贵金属催化剂的制备方法。
背景技术:
我国是煤炭生产大国,每年因煤炭的生产就会产出大量不同浓度的煤层气,开发有效的煤层气利用技术、减少甲烷的直接排放是我国建设节能环保型可持续发展模式、打造低碳经济体系的一个组成部分。结合节能减排以及对环境要求的提高,切实合理的开发煤层气这一低品位能源,并很好的将其转化为可利用的资源,扩大煤层气的使用范围和规模,提高煤层气的利用效率,具有节能和环保的双重意义,符合国家对能源政策的规划,符合国际环保组织对温室效应的控制,更符合我国对低品位能源开发使用的大力支持,促进国内对煤层气产业的快速发展。
煤层气开发利用的关键在于脱除其中的氧气,现有的煤层气脱氧技术主要有变压吸附分离法,焦炭燃烧法,催化脱氧法等。中国专利zl85103557公布了一种利用变压吸附法从煤层气中分离富集甲烷的方法。一般情况下,甲烷在浓缩提纯过程中排放废气的氧含量也被浓缩提高,由于废气中不可避免的含有5~15%的甲烷,致使排放的废气处于甲烷的爆炸极限范围,存在爆炸危险,这使得该技术的应用受到限制。
焦炭燃烧法脱氧(zl02113627.0、200610021720.1)是在高温条件下,富含甲烷气体中的氧与焦炭反应,同时部分甲烷与氧反应从而达到脱氧目的。其优点在于约70%的氧与焦炭反应,30%的氧与甲烷反应,因此甲烷损失较小。但缺点在于要消耗宝贵的焦炭资源,焦炭消耗成本约占整个运行费用的50%左右。此外,焦炭脱氧法在加焦、出渣时劳动强度大,环境灰尘大,难以实现自控操作及大规模生产,且焦炭中带有多种形态的硫化物,导致除氧后气体中含硫量增加。
催化脱氧工艺的本质是富燃贫氧气氛下甲烷的催化燃烧,在适当催化剂作用下,将ch4氧化转化为co2和h2o,此过程可将煤层气中含氧量降到0.5%以下,并彻底消除了操作过程的安全隐患。同时工艺操作简便,便于自动控制及大规模拓展,设备简单,从经济性角度而言,该技术亦具有较好的商业价值。催化脱氧根据催化剂活性组分可分为贵金属催化剂和非贵金属催化剂两大类。
国内外研究负载型贵金属催化剂的技术较为成熟。如中科院大连物化所在催化剂体系中添加具有储放氧功能的稀土铈组份,制备出新型负载型钯贵金属催化剂,可以将甲烷浓度为39.15%,氧气浓度为12.6%的煤层气脱氧处理后,产气中氧气浓度在0.1%以内,氧气转化率高于96%。由于贵金属催化剂价格昂贵且资源有限,应用范围受到限制。而非贵金属氧化物催化剂原料廉价易得,因而受到极大关注。但是非贵金属受活性所限,需要在较高的温度进行反应,能耗较大。
技术实现要素:
针对现有技术的不足,本发明提供了一种非贵金属催化剂的制备方法,在制备过程中引入配位剂和三元溶剂,通过配位、隔离作用,不仅避免了活性组分的流失,而且还极大促进了活性组分在催化剂上从内到外的均匀分布,有效提升了催化剂的反应性能。
一种非贵金属催化剂的制备方法,包括如下内容:
将mn、fe、co、ni、cu、zn、ce中的至少一种金属盐记为a和mg、ca、ti、zr、al中的至少一种金属盐记为b、配位剂、溶剂混合获得溶液c;
将溶液c进行晶化;
晶化后物料经洗涤、成型、干燥、焙烧后制得催化剂;
所述溶剂为水、乙醇、n,n-二甲基甲酰胺组成的三元溶剂,以溶剂的重量计:水为0.1~10wt%,乙醇为乙1~50wt%,余量为n,n-二甲基甲酰胺;优选水为1~5wt%,乙醇10~30wt%,余量为n,n-二甲基甲酰胺,所述金属盐a在溶液c中的质量分数为1~10wt%,优选1~5wt%,金属盐b在溶液c中的质量分数为20~70wt%,优选25~40wt%,配位剂在溶液c中的质量分数为1~20wt%,优选3~10wt%。
上述方法中,优选将mn、fe、cu、ce中的至少一种金属盐和zr、al中的至少一种金属盐、配位剂、溶剂混合获得溶液c。
上述方法中,所述金属盐为硝酸盐、硫酸盐、醋酸盐或氯化物中的一种或几种,优选为硝酸盐。
上述方法中,所述配位剂为草酸、琥珀酸、酒石酸、邻苯二甲酸、间苯二甲酸、对苯二甲酸、2,5-二羟基-对苯二甲酸、1,3,5-苯三羧酸、环戊烷四羧酸中的一种或几种,优选为2,5-二羟基-对苯二甲酸、1,3,5-苯三甲酸。
上述方法中,优选的混合过程如下:将mn、fe、co、ni、cu、zn、ce中的至少一种金属盐和mg、ca、ti、zr、al中的至少一种金属盐、加入溶剂中,调节溶液ph值为5~7后加入配位剂。
上述方法中,所述晶化温度为60~200℃,优选为80~150℃,所述晶化时间为1~48小时,优选为8~24小时。
上述方法中,将晶化所得物料用易挥发有机溶剂反复洗涤5~10次,过滤后,将所得滤饼进行成型处理后放入烘箱中干燥;然后将干燥后的成型体放入马弗炉在高温下焙烧,最后得到催化剂。所述易挥发有机溶剂为甲醇、乙醇、丙酮、二氯甲烷、氯仿中的一种或几种,优选为乙醇、丙酮。
上述方法中,所述干燥温度为60~200℃,优选为80~120℃;所述干燥时间为2~24小时,优选为8~24小时。
上述方法中,所述焙烧温度为300~800℃,优选为300~500℃;所述焙烧时间为4~24小时,优选为6~12小时。
本发明提供的一种非贵金属催化剂制备方法,其优势在于:
(1)本发明方法利用复合配位剂和三元溶剂的协同作用,将金属离子锚定在配位剂上,使其与金属离子通过配位作用形成大分子配合物;通过配合物的隔离作用,防止金属在载体上聚集,有效避免催化剂上出现游离物种,解决了活性组分的流失问题,防止了原料的浪费,最大限度提高了资源的利用率;通过复合配位剂发挥的配位、隔离、造孔等作用,极大促进了活性组分的均匀分布。tpr表征结果显示,表面物种和体相物种含量基本接近,表明本发明制备的催化剂上活性物种从内到外分布极为均匀;
(2)采用本发明方法制备的催化剂,不仅可用于煤层气脱氧中,对甲烷燃烧、天然气汽车尾气净化、vocs、co和h2等可燃气体亦具有较高的催化燃烧活性。
附图说明
图1是本发明实施例5制备的催化剂tpr谱图。
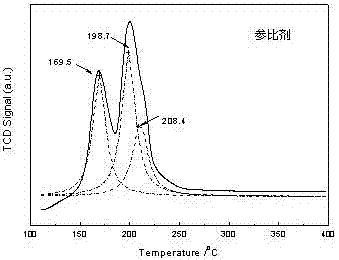
图2是本发明比较例1制备的参比剂tpr谱图。
图3是本发明实施例5制备的催化剂sem谱图。
图4是本发明比较例1制备的参比剂sem谱图。
具体实施方式
下面结合实施例进一步说明本发明的技术内容和效果,但不限制本发明。本发明中催化剂上的活性物种采用程序升温还原(tpr)进行表征,对tpr谱图进行分峰处理,获得代表不同活性物种的耗氢峰。根据tpr谱图中耗氢峰的位置可将活性物种分别归属为:游离物种(170℃附近),表面物种(198~202℃),体相物种(208~212℃)。其中,各活性物种的含量计算方法为:活性物种含量%=100*某物种的耗氢峰面积/总耗氢峰面积。
实施例1
按硝酸铜︰硝酸镁︰草酸︰2,5-二羟基对苯二甲酸︰三元溶剂(9.5wt%水-45.6wt%乙醇-44.9wt%n,n-二甲基甲酰胺)的摩尔比为0.008︰0.317︰0.034︰0.106︰1,首先称取硝酸铜和硝酸镁加入三元溶剂中,在45℃下搅拌使之完全溶解后,调节溶液ph值为6;然后,将草酸和2,5-二羟基对苯二甲酸作为复合配位剂加入溶液中,迅速搅拌使之混合均匀后,将该溶液转入内衬聚四氟乙烯的不锈钢反应釜中,于90℃下晶化8h。晶化产物用乙醇洗涤5次,抽滤后所得物料先成型,再于110℃下干燥12h。最后,将所得物料于300℃下焙烧10h,制得催化剂1#。
以煤层气脱氧为探针反应评价催化剂性能,原料气组成为:ch420vol%,o23vol%,余量为n2。反应温度为460℃,体积空速为10000h-1,待反应稳定后,在线色谱检测反应器出口的尾气中o2浓度为0.69%。
实施例2
按硝酸锰︰硝酸钙︰琥珀酸︰2,5-二羟基对苯二甲酸︰三元溶剂(7.8wt%水-10.5wt%乙醇-81.7wt%n,n-二甲基甲酰胺)的摩尔比为0.029︰0.229︰0.032︰0.076︰1,首先称取硝酸锰和硝酸钙加入三元溶剂中,在45℃下搅拌使之完全溶解后,调节溶液ph值为6;然后,将琥珀酸和2,5-二羟基对苯二甲酸作为复合配位剂加入溶液中,迅速搅拌使之混合均匀后,将该溶液转入内衬聚四氟乙烯的不锈钢反应釜中,于100℃下晶化12h。晶化产物用丙酮洗涤5次,抽滤后所得物料先成型,再于110℃下干燥12h。最后,将所得物料于300℃下焙烧8h,制得催化剂2#。
催化剂性能评价同实施例1,不同之处在于反应温度为450℃,反应器出口的尾气中o2浓度为0.73%。
实施例3
按硝酸铁︰硝酸铝︰酒石酸︰2,5-二羟基对苯二甲酸︰三元溶剂(6.2wt%水-35.5wt%乙醇-58.3wt%n,n-二甲基甲酰胺)的摩尔比为0.028︰0.195︰0.032︰0.065︰1,首先称取硝酸铁和硝酸铝加入三元溶剂中,在45℃下搅拌使之完全溶解后,调节溶液ph值为6;然后,将酒石酸和2,5-二羟基对苯二甲酸作为复合配位剂加入溶液中,迅速搅拌使之混合均匀后,将该溶液转入内衬聚四氟乙烯的不锈钢反应釜中,于110℃下晶化16h。晶化产物用乙醇洗涤5次,抽滤后所得物料先成型,再于110℃下干燥12h。最后,将所得物料于300℃下焙烧16h,制得催化剂3#。
催化剂性能评价同实施例1,不同之处在于反应温度为440℃,反应器出口的尾气中o2浓度为0.81%。
实施例4
按硝酸铈︰硝酸锆︰对苯二甲酸︰2,5-二羟基对苯二甲酸︰三元溶剂(1.2wt%水-40.2wt%乙醇-58.6wt%n,n-二甲基甲酰胺)的摩尔比为0.039︰0.189︰0.032︰0.063︰1,首先称取硝酸铈和硝酸锆加入三元溶剂中,在45℃下搅拌使之完全溶解后,调节溶液ph值为6;然后,将对苯二甲酸和2,5-二羟基对苯二甲酸作为复合配位剂加入溶液中,迅速搅拌使之混合均匀后,将该溶液转入内衬聚四氟乙烯的不锈钢反应釜中,于120℃下晶化20h。晶化产物用乙醇洗涤5次,抽滤后所得物料先成型,再于110℃下干燥12h。最后,将所得物料于300℃下焙烧12h,制得催化剂4#。
催化剂性能评价同实施例1,不同之处在于反应温度为430℃,反应器出口的尾气中o2浓度为0.58%。
实施例5
按硝酸铜︰硝酸铝︰1,3,5-苯三羧酸︰2,5-二羟基对苯二甲酸︰三元溶剂(5.4wt%水-26.7wt%乙醇-67.9wt%n,n-二甲基甲酰胺)的摩尔比为0.016︰0.121︰0.015︰0.033︰1,首先称取硝酸铜和硝酸铝加入三元溶剂中,在45℃下搅拌使之完全溶解后,调节溶液ph值为6;然后,将1,3,5-苯三羧酸和2,5-二羟基对苯二甲酸作为复合配位剂加入溶液中,迅速搅拌使之混合均匀后,将该溶液转入内衬聚四氟乙烯的不锈钢反应釜中,于120℃下晶化12h。晶化产物用乙醇洗涤5次,抽滤后所得物料先成型,再于110℃下干燥12h。最后,将所得物料于300℃下焙烧10h,制得催化剂5#。tpr表征结果显示,该催化剂上的表面铜含量为53%,体相铜含量为47%。
催化剂性能评价同实施例1,不同之处在于反应温度为420℃,色谱分析结果表明反应器出口的尾气中o2浓度为0,o2已完全转化。
实施例6
按氯化铜︰硝酸铝︰环戊烷四羧酸︰2,5-二羟基对苯二甲酸︰三元溶剂(3.5wt%水-20.9wt%乙醇-75.6wt%n,n-二甲基甲酰胺)的摩尔比为0.011︰0.098︰0.015︰0.033︰1,首先称取氯化铜和硝酸铝加入三元溶剂中,在45℃下搅拌使之完全溶解后,调节溶液ph值为6;然后,将环戊烷四羧酸和2,5-二羟基对苯二甲酸作为复合配位剂加入溶液中,迅速搅拌使之混合均匀后,将该溶液转入内衬聚四氟乙烯的不锈钢反应釜中,于100℃下晶化16h。晶化产物用乙醇洗涤5次,抽滤后所得物料先成型,再于110℃下干燥12h。最后,将所得物料于300℃下焙烧8h,制得催化剂6#。tpr表征结果显示,该催化剂上的表面铜含量为46%,体相铜含量为54%。
催化剂性能评价同实施例1,不同之处在于反应温度为410℃,反应器出口的尾气中o2浓度为0.26%。
实施例7
按醋酸铜︰硝酸铝︰邻苯二甲酸︰2,5-二羟基对苯二甲酸︰三元溶剂(5.1wt%水-16.1wt%乙醇-78.8wt%n,n-二甲基甲酰胺)的摩尔比为0.013︰0.087︰0.027︰0.029︰1,首先称取醋酸铜和硝酸铝加入三元溶剂中,在45℃下搅拌使之完全溶解后,调节溶液ph值为6;然后,将邻苯二甲酸和2,5-二羟基对苯二甲酸作为复合配位剂加入溶液中,迅速搅拌使之混合均匀后,将该溶液转入内衬聚四氟乙烯的不锈钢反应釜中,于110℃下晶化12h。晶化产物用乙醇洗涤5次,抽滤后所得物料先成型,再于110℃下干燥12h。最后,将所得物料于300℃下焙烧16h,制得催化剂7#。tpr表征结果显示,该催化剂上的表面铜含量为57%,体相铜含量为43%。
催化剂性能评价同实施例6,反应器出口的尾气中o2浓度为0.42%。
实施例8
按硫酸铜︰硝酸铝︰间苯二甲酸︰2,5-二羟基对苯二甲酸︰三元溶剂(2.2wt%水-30.5wt%乙醇-67.3wt%n,n-二甲基甲酰胺)的摩尔比为0.011︰0.075︰0.024︰0.025︰1,首先称取硫酸铜和硝酸铝加入三元溶剂中,在45℃下搅拌使之完全溶解后,调节溶液ph值为6;然后,将间苯二甲酸和2,5-二羟基对苯二甲酸作为复合配位剂加入溶液中,迅速搅拌使之混合均匀后,将该溶液转入内衬聚四氟乙烯的不锈钢反应釜中,于110℃下晶化12h。晶化产物用乙醇洗涤5次,抽滤后所得物料先成型,再于110℃下干燥12h。最后,将所得物料于300℃下焙烧12h,制得催化剂8#。tpr表征结果显示,该催化剂上的表面铜含量为39%,体相铜含量为61%。
催化剂性能评价同实施例6,反应器出口的尾气中o2浓度为0.35%。
实施例9
催化剂制备方法同实施例5,不同之处在于焙烧温度为400℃,制得催化剂9#。tpr表征结果显示,该催化剂上的表面铜含量为54%,体相铜含量为46%。
催化剂性能评价同实施例5,色谱分析结果表明反应器出口的尾气中o2浓度为0,o2已完全转化。
实施例10
催化剂制备方法同实施例5,不同之处在于焙烧温度为450℃,制得催化剂10#。tpr表征结果显示,该催化剂上的表面铜含量为51%,体相铜含量为49%。
催化剂性能评价同实施例5,色谱分析结果表明反应器出口的尾气中o2浓度为0,o2已完全转化。
实施例11
催化剂制备方法同实施例5,不同之处在于焙烧温度为500℃,制得催化剂11#。tpr表征结果显示,该催化剂上的表面铜含量为48%,体相铜含量为52%。
催化剂性能评价同实施例5,色谱分析结果表明反应器出口的尾气中o2浓度为0,o2已完全转化。
实施例12
催化剂制备方法同实施例5,不同之处在于焙烧温度为550℃,制得催化剂12#。tpr表征结果显示,该催化剂上的表面铜含量为29%,体相铜含量为71%。
催化剂性能评价同实施例5,色谱分析结果表明反应器出口的尾气中o2浓度为0,o2已完全转化。
比较例1
采用常规浸渍法制备负载型氧化铜催化剂,具体操作为:称取硝酸铜100g,溶于水中,加入氧化铝载体160g,进行等体积浸渍。室温下静置8h后,放入烘箱于120℃下处理12h,然后在500℃下焙烧10h,制得参比剂。tpr表征结果显示,参比剂上的游离铜含量为34%,表面铜含量为41%,体相铜含量为25%。
催化剂性能评价同实施例5,反应器出口的尾气中o2浓度为1.52%。
实施例5制备的催化剂5#与比较例1制备的参比剂对比结果表明,采用常规浸渍法制备的负载型氧化铜催化剂,含有游离铜、表面铜、体相铜三类物种,其中游离铜在反应过程中易于脱落,在参比剂中含量为34%,这表明有三分之一的铜物种被浪费掉。而采用本发明方法制备的催化剂,在tpr谱图中只有表面铜和体相铜两类物种,这说明该方法能将铜物种有效保留在催化剂中,避免了浪费。与此同时,从表面铜与体相铜的含量近似相等也可以看出,该催化剂上铜物种从内到外均匀分布。该催化剂的比表面积(240m2/g)也远高于参比剂(190m2/g),这说明催化剂上的孔道结构也比参比剂发达,能更有效的利用铜物种。因此,具有较高的活性和稳定性。