一种硫掺杂氮化碳光催化剂及其制备方法、应用与流程
本发明涉及光催化剂领域,特别涉及一种硫掺杂氮化碳光催化剂及其制备方法、应用。
背景技术:
随着能源危机日益加重,新型能源的开发引起了世界范围内的重视,其中如何高效利用太阳能一直是人们研究的热点。在过去几十年中,不同种类的光催化剂得到人们的广泛使用,如金属氧化物、硫化物和氮化物(TiO2、ZnO、Fe2O3、SnO2、WO3、CdS)等,其中TiO2以其活性高、热稳定性好、持续性长、价格便宜、对人体无害等特征倍受人们青睐。由于TiO2对很多有机污染物的光催化氧化活性高,因此它在环境污染治理方面也扮演着极其重要的角色,被广泛用于光催化处理多种有机物。但由于TiO2的光生电子和空穴容易发生复合,光催化效率低,带隙较宽(约3.2eV),只能在紫外区显示光化学活性,对太阳能的利用率不到5%。因此,如何提高太阳光的利用率成为近年的研究热点,除了改进技术提高原有的光催化剂性能之外,一些新型光催化剂也广受关注。近些年,石墨化氮化碳(g-C3N4)由于其类碳结构及良好的光催化性能而引起人们的广泛关注。g-C3N4是一种有机半导体材料,禁带宽度约2.7eV,具有合适的导带价带位置,从骨架的拓扑结构可看出,理想的g-C3N4应是N连接的C3N3芳香环,类似石墨烯,形成二维共轭的平面结构,从XRD、SEM、TEM中可验证此种结构。其独特的结构和高的聚合程度使其具有较高的热稳定性和化学稳定性(酸、碱、有机溶剂)以及良好的电子结构。
目前制备g-C3N4光催化剂的方法主要采用的是高温煅烧热缩聚法,即将前驱体加热至500~600℃,发生缩聚反应得到产物。此方法得到的g-C3N4比表面积小,对光的吸收降低,因此光催化性能往往不理想。
技术实现要素:
为了克服氮化碳在现有技术上存在的比表面积小,可见光利用率较低等不足,本发明的目的在于提供一种高比表面积硫掺杂氮化碳光催化剂的制备方法,制备得到的硫掺杂氮化碳光催化剂具有更大的比表面积,更强光吸收及更优越的光催化活性。
本发明的另一目的在于提供上述制备方法得到的硫掺杂氮化碳光催化剂。
本发明的再一目的在于提供上述硫掺杂氮化碳光催化剂的应用。
本发明的目的通过以下技术方案实现:
一种硫掺杂氮化碳光催化剂的制备方法,包括以下步骤:
S1将硫脲于500~650℃中煅烧,保温时间为2~6小时,得到硫掺杂石墨相氮化碳;
S2将S1得到的硫掺杂石墨相氮化碳研磨成粉,于400~550℃再次煅烧,保温1~4小时。
步骤S1所述的煅烧,升温速率为1~5℃/min。
步骤S2所述的煅烧,升温速率为2~10℃/min。
步骤S1所述的煅烧,具体为:
将硫脲盛于坩埚中加盖,置于马弗炉中,于500~600℃煅烧,保温时间为3~5小时,升温速率为2~4℃/min。
步骤S2所述的煅烧,具体为:
将硫脲盛于坩埚中不加盖,并铺展开,于450~550℃煅烧,升温速率为4~7℃/min。
所述硫掺杂氮化碳光催化剂的制备方法制备得到的硫掺杂氮化碳光催化剂。
所述的硫掺杂氮化碳光催化剂的应用,用于对苯胺溶液进行光降解。
本发明的原理为:
本发明首先将硫脲通过高温煅烧缩聚形成硫掺杂氮化碳,冷却后再次煅烧,利用空气中的氧气使其剥离氧化而制备得到高比表面积、高催化活性的硫掺杂氮化碳。
与现有技术相比,本发明具有以下优点和有益效果:
(1)本发明制备得到的掺杂氮化碳光催化剂,具有更大的比表面积(191.2m2/g),可提供更多的反应位点,使其在可见光的照射下对有机污染物具有较强的光催化降解能力。
(2)本发明制备得到的硫掺杂氮化碳光催化剂,对太阳光具有更强的光吸收。
(3)本发明的制备方法,步骤操作简单,催化剂活性高。
(4)本发明制备得到的掺杂氮化碳光催化剂,用于光催化降解苯胺溶液,在1小时内可完全降解苯胺。
附图说明
图1是本发明的实施例1制得的CN-T-2的C元素的XPS光谱图。
图2是本发明的实施例1制得的CN-T-2的O元素的XPS光谱图。
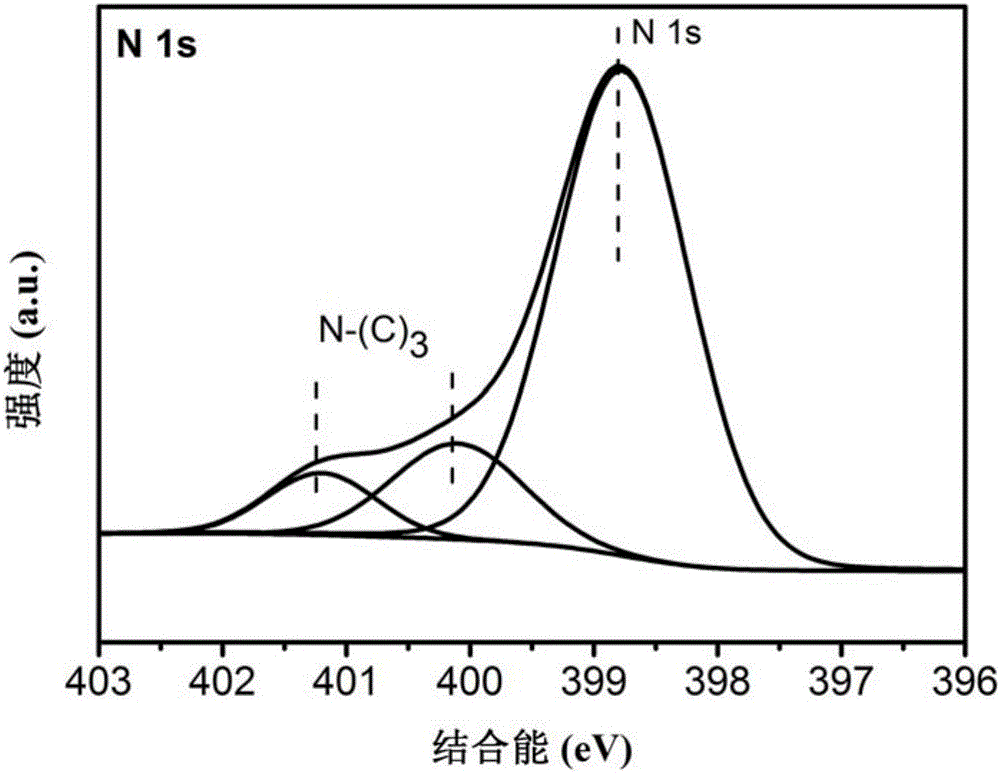
图3是本发明的实施例1制得的CN-T-2的N元素的XPS光谱图。
图4是本发明的实施例1制得的CN-T-2的S元素的XPS光谱图。
图5是本发明的实施例1制备的CN-T-1、CN-T-2及CN-D-1、CN-D-2的DRS图。
图6是本发明的实施例1制备的CN-T-1、CN-T-2的XRD图。
图7本发明的各实施例中制备得到的光催化剂对苯胺溶液进行光降解的降解效果对比图。
具体实施方式
下面结合实施例,对本发明作进一步地详细说明,但本发明的实施方式不限于此。
实施例1
将10g硫脲置于坩埚中加盖,在马弗炉中煅烧,煅烧温度为550℃,保温时间为4小时,升温速率为2.3℃/min;自然冷却后研磨成粉,得到硫掺杂石墨相氮化碳(CN-T-1),称取1.2g盛于坩埚中不加盖,并铺展开,再次煅烧,煅烧温度为500℃,保温时间为2小时,升温速率为5℃/min。自然冷却后得到高比表面积改性氮化碳光催化剂(CN-T-2),产率约为12%。
将实施例1中制备得到的光催化剂进行XPS、DRS、XRD等表征,结果如图1~7所示。
结论:图1~4所示分别是CN-T-2的C、O、N、S的XPS图,从图中看到C、O、N的成分及价态与氮化碳一致,另外还检测到S元素的存在。
图5所示分别是CN-T-1、CN-T-2的DRS对比图,从图中可以看出CN-T-2相对CN-T-1发生了蓝移,说明了煅烧使得CN-T-2具有更多的微观片层及更细的微观尺寸,而相比使用二氢二胺制备的不含硫元素的氮化碳(一次煅烧CN-D-1及再次煅烧CN-D-2),含S元素的氮化碳CN-T-1、CN-T-2则有相对较强的吸光度,证明了S元素的加入可以调整氮化碳的禁带宽度,增强对可将光的吸收。
图6所示分别是CN-T-1、CN-T-2的XRD对比图。从图中可以看出,两种物质的出峰情况较好,没有杂峰出现。CN-T-2在27°及13°附近的峰相比CN-T-1都有所减弱,说明煅烧使得CN-T-2结晶度有所降低,形成更多微观片层结构;同时CN-T-2在27°左右的峰位置相对CN-T-1有所偏移,说明煅烧后CN-T-2的层间距更大。
煅烧后的CN-T-2较蓬松,通过BET比表面积测试发现,CN-T-2的比表面积达到191.2m2/g,而CN-T-1则为则为32.53m2/g,比表面积有了较大的提升。
实施例2
将10g硫脲置于坩埚中加盖,在马弗炉中煅烧,煅烧温度为550℃,保温时间为4小时,升温速率为2.3℃/min;自然冷却后研磨成粉,称取1.2g盛于坩埚中不加盖,并铺展开,再次煅烧,煅烧温度为500℃,保温时间为1.0小时,升温速率为5℃/min。自然冷却后得到高比表面积改性氮化碳光催化剂(CN-T-21小时),产率约为18%。
实施例3
将10g硫脲置于坩埚中加盖,在马弗炉中煅烧,煅烧温度为550℃,保温时间为4小时,升温速率为2.3℃/min;自然冷却后研磨成粉,称取1.2g盛于坩埚中不加盖,并铺展开,再次煅烧,煅烧温度为500℃,保温时间为3小时,升温速率为5℃/min。自然冷却后得到高比表面积改性氮化碳光催化剂(CN-T-23小时),产率约为8%。
实施例4
将10g硫脲置于坩埚中加盖,在马弗炉中煅烧,煅烧温度为550℃,保温时间为4小时,升温速率为2.3℃/min;自然冷却后研磨成粉,称取1.2g盛于坩埚中不加盖,并铺展开,再次煅烧,煅烧温度为500℃,保温时间为4小时,升温速率为5℃/min。自然冷却后得到高比表面积改性氮化碳光催化剂(CN-T-24小时),产率约为4%。
实施例5
将10g硫脲置于坩埚中加盖,在马弗炉中煅烧,煅烧温度为650℃,保温时间为2小时,升温速率为5℃/min;自然冷却后研磨成粉,称取1.2g盛于坩埚中不加盖,并铺展开,再次煅烧,煅烧温度为550℃,保温时间为1小时,升温速率为10℃/min。自然冷却后得到高比表面积改性氮化碳光催化剂,产率约为15%。
实施例6
将10g硫脲置于坩埚中加盖,在马弗炉中煅烧,煅烧温度为500℃,保温时间为6小时,升温速率为1℃/min;自然冷却后研磨成粉,称取1.2g盛于坩埚中不加盖,并铺展开,再次煅烧,煅烧温度为450℃,保温时间为4小时,升温速率为2℃/min。自然冷却后得到高比表面积改性氮化碳光催化剂,产率约为16%。
实施例7
将10g硫脲置于坩埚中加盖,在马弗炉中煅烧,煅烧温度为600℃,保温时间为3小时,升温速率为4℃/min;自然冷却后研磨成粉,称取1.2g盛于坩埚中不加盖,并铺展开,再次煅烧,煅烧温度为500℃,保温时间为2小时,升温速率为7℃/min。自然冷却后得到高比表面积改性氮化碳光催化剂,产率约为10%。
对苯胺的光催化降解性能验证:
向光反应器中加入150ml浓度为10mg/L的苯胺溶液,取50mg上述实施例制得的CN-T-1、CN-T-2投入其中,暗反应30min后达到吸附平衡,然后在300W氙灯下照射,每15min取一次样,再用紫外-可见分光光度计测量溶液的吸光度并计算剩余苯胺含量,降解结果见图7。从图中可以看出,经过两次煅烧的氮化碳降解苯胺效果都有所提升,其中CN-T-2 3小时的降解效果最好,经过60min氙灯照射后可将苯胺完全降解,而CN-T-1则约为70%,提升效果明显。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受所述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!