一种磁性高分子吸附材料、制备方法和应用与流程
本发明属于高分子材料制备方法及其应用领域,具体涉及一种磁性高分子吸附材料及其制备方法与应用。
背景技术:
近年来,磁性高分子聚合材料因其具备的较高吸附容量以及良好分离性能,在环境污染物前处理以及吸附去除等方面显示了巨大的应用潜力。目前应用于环境领域的磁性高分子吸附材料主要分为两大类,一类是离子交换型,另一类为具有高比表面积的超高交联材料。
经检索,现有技术已经公布了相关的申请案;中国专利号cn201010017687.1,公开日为2012.07.04的申请案公开了一种磁性丙烯酸系强碱阴离子交换树脂的制备方法,该申请案中的磁性树脂对水体中溶解性有机物、硝酸盐、磷酸盐等阴离子有较好的去除效果;中国专利号cn201010500161.9,公开日为2011.01.19的申请案公开了一种磁性弱酸系阳离子交换树脂的合成方法,该申请案中的磁性树脂主要用于氨氮和金属等阳离子物质的去除,应用范围有一定的局限性。
中国专利号cn201110426866.5,公开日为2012.06.27的申请案公开了一种高比表面积磁性微球树脂及其制备方法和应用,该申请案中的材料对水体中的非极性以及电中性物质有非常优异的去除效率,然而对强极性或带电的物质的去除效果并不理想。
基于现有技术的缺陷,中国专利号cn201310106265.5,公开日为2013.06.19的申请案公开了一种磁性胺基修饰超高交联树脂及其制备方法;中国专利号cn201310330339.3,公开日为2014.01.08的申请案公开了一种弱酸修饰高比表面积磁性树脂、其制备方法及其高效净化微污染水体的方法;上述两个申请案中开发的磁性材料均属于基团修饰的超高交联材料,这类材料既有丰富的孔道,又含有较高的离子交换能力,有效地扩大了磁性高分子材料的应用范围。然而这类材料也存在不可避免的缺陷:1)很难做到同时具备很高的比表面积以及离子交换能力;2)需要针对目标物质修饰特定基团,难以适应对水体中有机物的广谱性吸附。
经检索,中国专利号cn201510603600.1,公开日为2015.11.11的申请案公开了一种亲水性聚合物微球及其制备方法,该方法制备的材料对于亲水性和疏水性污染物都有较好的吸附效果,该申请案的方法以n-乙烯吡咯烷酮、亲油性的二乙烯苯为主要单体,按比例将n-乙烯吡咯烷酮、二乙烯苯和引发剂、致孔剂混合均匀,搅拌、升温反应,反应结束后进行沉淀过滤、洗涤、再过滤和干燥,得到亲水性聚合物微球;然而由于n-乙烯基吡咯烷酮是一种可与水完全互溶的高分子化合物,而二乙烯苯具有较强的疏水性,在聚合过程中两者难以很好结合,利用该申请案的制备方法合成出的亲水性聚合物微球整体产率不稳定,需要通过提高n-乙烯基吡咯烷酮的用量来提高亲水性聚合物微球的亲水性和比表面积,不具备大规模推广价值;且制备出的材料不具备磁性,应用范围较为局限,无法适应对水体中有机物的广谱性吸附。
因此,如何制备具有一定磁性,同时具有高比表面积的分子吸附材料以适用于水体中有机物的广谱性吸附是磁性高分子材料制备过程中需要继续突破的难题。
技术实现要素:
1.要解决的问题
针对现有技术中制备的磁性材料吸附具有局限性,不能适应对水体中有机物的广谱性吸附的缺陷,本发明旨在提供一种粒径均匀性好、磁性强、吸附性强的磁性高分子吸附材料。
2.技术方案
为了实现以上目的,本发明采用如下技术方案:
本发明提供了一种磁性高分子吸附材料的制备方法,包括以下步骤:
(1)、制备磁性纳米粒子;
(2)、配制油相溶液:将步骤(1)中制备的磁性纳米粒子溶解于致孔剂中,分别加入n-乙烯吡咯烷酮、二乙烯苯和引发剂,在冰浴条件下使其混合均匀;
(3)、合成磁性高分子吸附材料:在水溶液中加入乳化剂、分散剂;在低于60℃条件下,加入一部分步骤(2)中配制的油相溶液,温度升至60℃以上时加入其余油相溶液,每次加入的油相溶液占总油相溶液体积分数的10~90%,搅拌反应,再将反应后溶液进行沉淀、过滤、洗涤、干燥,最终得到磁性高分子吸附材料。
作为本发明更进一步的改进,所述的磁性纳米粒子为fe3o4@有机酸纳米粒子,其制备过程为:将含有fe2+和fe3+的可溶性盐配成溶液,混合、通氮气保护,在60℃~100℃温度下加入沉淀剂、有机酸,反应0.5~12小时后将溶液ph值调为酸性,将产物洗涤、干燥后即可得到磁性fe3o4@有机酸纳米粒子。
作为本发明更进一步的改进,所述步骤(3)中搅拌速率为:100~1500rpm,搅拌时间为:12~80h,搅拌反应的温度为60~95℃。
作为本发明更进一步的改进,所述的n-乙烯吡咯烷酮的质量占n-乙烯吡咯烷酮和二乙烯苯总质量的10~90%。
作为本发明更进一步的改进,所述的致孔剂体积为水溶液体积的0.05~2倍;所述的乳化剂质量占n-乙烯吡咯烷酮和二乙烯苯总质量的0.1~20%;所述的分散剂质量占n-乙烯吡咯烷酮和二乙烯苯总质量的0~10%;所述引发剂质量占n-乙烯吡咯烷酮、二乙烯苯总质量的0.1~5%。
作为本发明更进一步的改进,引发剂为偶氮类或苯甲酰类;致孔剂为甲醇、甲苯、环己醇、dmf、dmso中的一种或几种;乳化剂为阴离子表面活性剂;分散剂为pvp、hec、peg中的一种或几种;所述的fe2+和fe3+的可溶性盐的摩尔比为1:(0.23~5.5);所述的有机酸质量为磁性fe3o4纳米粒子的0.5~5倍;沉淀剂为碱溶液。
作为本发明更进一步的改进,由所述的磁性高分子吸附材料的制备方法合成的磁性高分子吸附材料,所述材料平均粒径为2~100μm,磁化强度为5~19.5emu/g,比表面积为210~950m2/g。
作为本发明更进一步的改进,所述材料应用于吸附溶液中的无机物及有机物。
作为本发明更进一步的改进,所述材料应用于无机物及有机物的控制释放。
作为本发明更进一步的改进,所述材料应用于不同物质的分离。
3.有益效果
相比于现有技术,本发明的有益效果为:
(1)本发明的磁性高分子吸附材料的制备方法,制备出适用于水体中有机物的广谱性吸附材料,该方法首先配制油相溶液:将制备的磁性纳米粒子溶解于致孔剂中,分别加入n-乙烯吡咯烷酮、二乙烯苯和引发剂,在低温下使其混合均匀;然后在水溶液中加入乳化剂、分散剂;在低于60℃条件下,加入一部分油相溶液,温度升至60℃以上时加入其余油相溶液,搅拌反应,将反应后溶液进行沉淀、过滤、洗涤、干燥,最终得到磁性高分子吸附材料;本发明的方法克服了现有技术以n-乙烯吡咯烷酮和二乙烯苯为单体制备吸附材料过程中单纯采用将n-乙烯吡咯烷酮、二乙烯苯、引发剂、致孔剂混合均匀,进行搅拌、升温反应时n-乙烯吡咯烷酮的强亲水性和二乙烯苯强疏水性导致二者结合性不好,制备的材料产率不稳定的缺陷。
(2)本发明的磁性高分子吸附材料的制备方法,克服了现有技术中以n-乙烯吡咯烷酮和二乙烯苯为单体制备吸附材料过程中磁性纳米粒子难以引入,制备出的材料具有一定的吸附性能,并不具备磁性的缺陷;将n-乙烯吡咯烷酮、二乙烯苯、磁性纳米粒子三种性质差别较大的物质结合,在简单的制备步骤中将磁性纳米粒子引入,制备出的材料既具有较强的磁性,又具有较大的比表面积,吸附性能和应用范围均得到大幅度提高。
(3)本发明的磁性高分子吸附材料的制备方法,所需原料成本低廉,制备方法简单,利于推广。
(4)本发明的磁性高分子吸附材料,其平均粒径在2~100μm之间,粒径均一性较好;磁化强度在5~19.5emu/g,比表面积210~950m2/g,不仅具有较大的比表面积,而且具有较强的磁性,对亲水性、疏水性物质、金属离子等都有很好的吸附效果,对比相同比表面积的oasishlb材料和xad-4树脂材料,本发明的材料对污染物吸附量有显著提高。
(5)本发明的磁性高分子吸附材料,其在外加磁场的作用下,可从溶液中迅速分离出来,操作简单,有效的减少了分析时间。
(6)本发明的磁性高分子吸附材料,具有环境友好、可进行大量生产、反复使用等优点,在环境治理、检验检测、药物分离等领域应用前景广阔。
附图说明
图1为本发明的磁性高分子吸附材料的基本结构示意图;
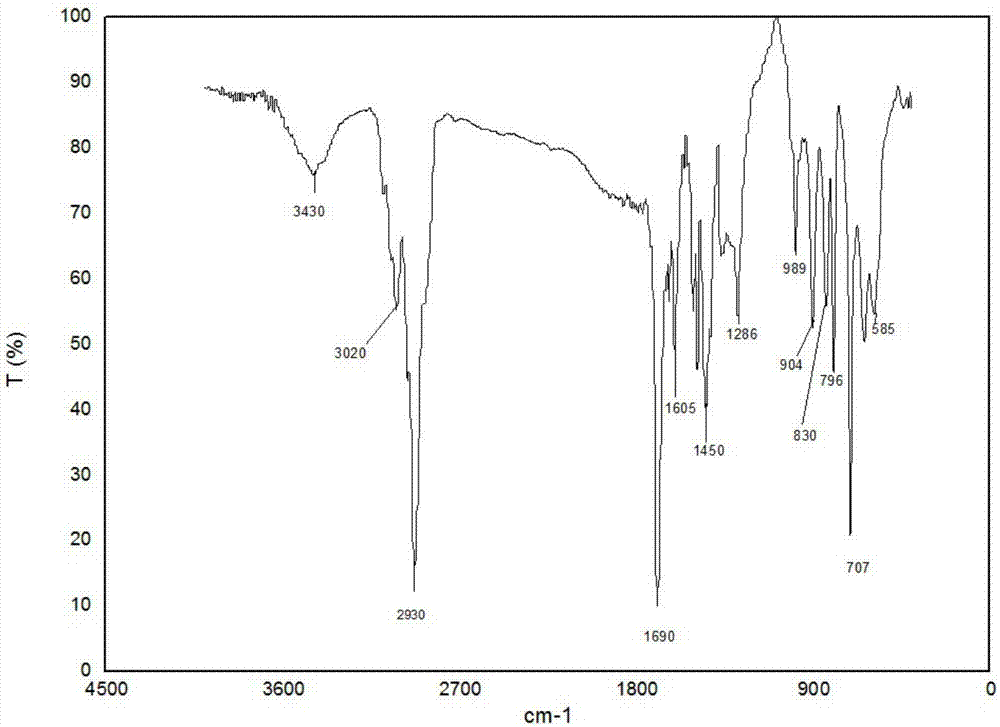
图2为本发明的磁性高分子吸附材料的ft-ir图谱;
图3为本发明的磁性高分子吸附材料的sem图谱。
具体实施方式
下面结合具体实施例对本发明进一步进行描述。
具体实施方式
下面结合具体实施例对本发明做进一步描述,以下实施例仅用来更好的说明技术方案,而不能以此限制本发明的保护范围。
实施例1
本实施例中制备磁性高分子吸附材料的步骤如下:
(1)、制备磁性fe3o4@油酸纳米粒子:在250ml的烧瓶中加入1g氯化亚铁和2g氯化铁、100ml水,电动搅拌混合均匀,通氮气保护,然后在60℃温度下加入4mol/l的氢氧化钠溶液20ml、油酸0.75g,此时,所述的油酸的质量为磁性fe3o4纳米粒子的0.5倍。反应10小时后将溶液ph值调为3,将产物用水和乙醇洗涤5次,干燥后即可得到磁性fe3o4@油酸纳米粒子。
(2)、配制油相溶液:将2g磁性fe3o4@油酸纳米粒子溶解于30ml甲苯溶液中,分别加入3gn-乙烯吡咯烷酮、3g二乙烯苯、0.05gaibn,冰浴混合均匀得到油相溶液,所述的n-乙烯吡咯烷酮质量占n-乙烯吡咯烷酮和二乙烯苯总质量的50%,引发剂aibn占n-乙烯吡咯烷酮和二乙烯苯总质量的0.8%。
(3)、合成磁性高分子吸附材料:在50ml水溶液中加入0.5g乳化剂sds、0.25g分散剂hec;在45℃下加一半上述油相溶液,在65℃下加另一半油相溶液,所述的乳化剂质量占n-乙烯吡咯烷酮和二乙烯苯总质量的8.3%,分散剂质量占n-乙烯吡咯烷酮和二乙烯苯总质量的4.2%,300rpm、65℃搅拌24小时后,将反应后溶液进行沉淀、过滤,甲醇洗涤5次,65℃下干燥12小时,最终得到磁性高分子吸附材料。
该材料基本结构如图1所示,根据反应机理进行推导,所加单体二乙烯苯的两个双键都可能打开进行链增长;反应中被活化的单体自由基可以与n-乙烯吡咯烷酮、二乙烯苯或者含有油酸的磁性纳米粒子形成新的自由基进行链增长直至链终止。
对该材料进行的红外光谱(ft-ir)的结构表征图谱如图2所示,1690cm-1的特征峰为n-乙烯吡咯烷酮的c=o键,707cm-1和796cm-1是苯环的特征峰,3430cm-1和585cm-1是fe3o4纳米粒子的特征峰。
对该材料进行的扫描电子显微镜(sem)扫描图谱如图3所示,该磁性高分子吸附的粒径集中分布在100μm,由图1可知该材料的粒径分布较为均匀,经全自动比表面积和孔隙度分析仪测定,其比表面积为681.5m2/g;经震动样品磁强计测定,其磁化强度为15emu/g;试验证明,该磁性高分子吸附材料在25℃条件下对水中硝基苯类化合物的吸附量达到389mg/g。
waters公司商业化生产的相同比表面积的oasishlb材料的吸附量只有157mg/g,商业化的xad-4树脂的吸附量只有25.7mg/g,本实施例制备的材料对污染物吸附量有显著提高。吸附实验的后处理只需外加磁铁即可将吸附材料与溶液分离;对比商业化的oasishlb和xad-4树脂,本实施例制备的材料吸附后处理过程简单易行,非常便捷。
对比例a
(1)、制备磁性fe3o4@油酸纳米粒子:在250ml的烧瓶中加入1g氯化亚铁和2g氯化铁、100ml水,电动搅拌混合均匀,通氮气保护,然后在60℃温度下加入4mol/l的氢氧化钠溶液20ml、油酸4g,此时,所述的油酸的质量为磁性fe3o4纳米粒子的0.9倍。反应10小时后将溶液ph值调为3。将产物用水和乙醇洗涤5次、干燥后即可得到磁性fe3o4@油酸纳米粒子。
(2)、将n-乙烯吡咯烷酮、二乙烯苯,致孔剂甲苯和引发剂aibn混合后加入含有无水乙醇有机相的三口烧瓶中,然后加入步骤(1)中的磁性fe3o4@油酸纳米粒子,升温反应一定时间之后,过滤洗涤干燥产物。
经检测,根据上述步骤制得材料磁强度很小(<0.1emu/g),不能在溶液中进行磁分离。多次改变加入磁性纳米粒子的方式以及加入的比例都不能合成磁性高分子吸附材料。
对比例b
(1)、制备磁性fe3o4@油酸纳米粒子:在250ml的烧瓶中加入1g氯化亚铁和2g氯化铁、100ml水,电动搅拌混合均匀,通氮气保护,然后在60℃温度下加入4mol/l的氢氧化钠溶液20ml、油酸4g,此时,所述的油酸的质量为磁性fe3o4纳米粒子的0.9倍。反应10小时后将溶液ph值调为3。将产物用水和乙醇洗涤5次、干燥后即可得到磁性fe3o4@油酸纳米粒子。
(2)、将n-乙烯吡咯烷酮、二乙烯苯,致孔剂甲苯和引发剂aibn混合后加入含有氯化钠、明胶、聚乙烯醇、表面活性剂中一种或者几种的水溶液中,然后加入步骤(1)中的磁性fe3o4@油酸纳米粒子,升温反应一定时间之后,过滤洗涤干燥产物。
经检测,根据上述步骤制得材料磁强度很小(<0.3emu/g),不能在溶液中进行磁分离。多次改变加入磁性纳米粒子的方式也都不能合成磁性高分子吸附材料。
由对比例a和对比例b可知,按照现有技术以n-乙烯吡咯烷酮、二乙烯苯为单体的制备方法,磁性纳米粒子引入难度较大,难以制得具有磁性的高分子吸附材料。
实施例2
本实施例中制备磁性高分子吸附材料的步骤如下:
(1)、制备磁性fe3o4@油酸纳米粒子:在500ml的烧瓶中加入2.6g氯化亚铁和6.4氯化铁、200ml水,电动搅拌混合均匀,通氮气保护,然后在100℃温度下加入2mol/l的氢氧化钾溶液40ml、油酸4g,所述的油酸的质量为磁性fe3o4纳米粒子的2倍;反应6小时后将溶液ph值调为3。将产物用水和甲醇洗涤5次、干燥后即可得到磁性fe3o4@油酸纳米粒子。
(2)、配制油相溶液:将1g磁性fe3o4@油酸纳米粒子溶解于10ml环己醇溶液中,分别加入2gn-乙烯吡咯烷酮、1.5g二乙烯苯、0.07gaibn,冰浴混合均匀得到油相溶液。所述的n-乙烯吡咯烷酮质量占n-乙烯吡咯烷酮和二乙烯苯总质量的57%,引发剂aibn占n-乙烯吡咯烷酮和二乙烯苯总质量的2%。
(3)、合成磁性高分子吸附材料:在200ml水溶液中加入0.1g乳化剂sds、0.05g分散剂hec;在45℃下加10%上述油相溶液油相溶液,在65℃下加入其余上述油相溶液,所述的乳化剂质量占n-乙烯吡咯烷酮和二乙烯苯总质量的2.9%,分散剂质量占n-乙烯吡咯烷酮和二乙烯苯总质量的1.45%,500rpm、65℃条件下搅拌48小时后,将反应后溶液进行沉淀、过滤,甲醇洗涤5次,75℃下干燥24小时,最终得到磁性高分子吸附材料。
本实施例中所得磁性高分子吸附材料的粒径集中分布在10μm,比表面积为714m2/g,磁化强度为12emu/g。该材料在25℃条件下对水中四环素的吸附量达到275mg/g,将已经吸四环素饱和的磁性高分子材料外加磁场进行分离后加入纯水中进行超声,可调节超声功率和时间来控制释放在水体中的四环素含量,因此,该材料还可用于四环素的控制释放。
实施例3
本实施例中制备磁性高分子吸附材料的步骤如下:
(1)、制备磁性fe3o4@油酸纳米粒子:在250ml的烧瓶中加入1g氯化亚铁和7g氯化铁、100ml水,电动搅拌混合均匀,通氮气保护,然后在70℃温度下加入饱和氨水溶液20ml、油酸3g,所述的油酸的质量为磁性fe3o4纳米粒子的1.7倍;反应0.5小时后将溶液ph值调为3。将产物用水和甲醇洗涤5次、干燥后即可得到磁性fe3o4@油酸纳米粒子。
(2)、配制油相溶液:将2g磁性fe3o4@油酸纳米粒子溶解于30mldmf液中,分别加入3gn-乙烯吡咯烷酮、2g二乙烯苯、0.1gbpo,冰浴混合均匀得到油相溶液。所述的n-乙烯吡咯烷酮质量占n-乙烯吡咯烷酮和二乙烯苯总质量的60%,引发剂bpo占n-乙烯吡咯烷酮和二乙烯苯总质量的2%。
(3)、合成磁性高分子吸附材料:在100ml水溶液中加入0.005g乳化剂sds、0.5g分散剂pvp;在55℃下加一半上述油相溶液油相溶液,在75℃下加另一半上述油相溶液,所述的乳化剂质量占n-乙烯吡咯烷酮和二乙烯苯总质量的0.1%,分散剂质量占n-乙烯吡咯烷酮和二乙烯苯总质量的10%,300rpm、75℃搅拌24小时后,将反应后溶液进行沉淀、过滤,甲醇洗涤5次,65℃下干燥12小时,最终得到磁性高分子吸附材料。
本实施例中所得磁性高分子吸附材料的粒径集中分布在120μm,比表面积为254m2/g,磁化强度为9emu/g。该材料在25℃条件下对水中铜离子的吸附量达到75mg/g,将吸附铜离子饱和的磁性高分子材料外加磁场分离后加入0.1mol/l的盐酸溶液中,可以将铜离子完全释放,因此,该材料还可用于铜离子的控制释放。
实施例4
本实施例中制备磁性高分子吸附材料的步骤如下:
(1)、制备磁性fe3o4@油酸纳米粒子:在500ml的烧瓶中加入3g氯化亚铁和8g氯化铁、100ml水,电动搅拌混合均匀,通氮气保护,然后在80℃温度下加入4mol/l的氢氧化钠溶液20ml、油酸7g,所述的油酸的质量为磁性fe3o4纳米粒子的1.2倍;反应6小时后将溶液ph值调为3。将产物用水和乙醇洗涤5次、干燥后即可得到磁性fe3o4@油酸纳米粒子。
(2)、配制油相溶液:将2g磁性fe3o4@油酸纳米粒子溶解于60mldmso溶液中,分别加入2gn-乙烯吡咯烷酮、4g二乙烯苯、0.01gbpo,冰浴混合均匀得到油相溶液。所述的n-乙烯吡咯烷酮质量占n-乙烯吡咯烷酮和二乙烯苯总质量的33%,引发剂bpo占n-乙烯吡咯烷酮和二乙烯苯总质量的0.2%。
(3)、合成磁性高分子吸附材料:在30ml水溶液中加入0.5g乳化剂sds、0.2g分散剂peg、0.075g分散剂hec;在40℃下加90%上述油相溶液油相溶液,在64℃下加入其余上述油相溶液,所述的乳化剂质量占n-乙烯吡咯烷酮和二乙烯苯总质量的8.3%,分散剂质量占n-乙烯吡咯烷酮和二乙烯苯总质量的4.6%;800rpm、64℃搅拌12小时后,将反应后溶液进行沉淀、过滤,甲醇洗涤5次,65℃下干燥12小时,最终得到磁性高分子吸附材料。
本实施例中所得磁性高分子吸附材料的粒径集中分布在70μm,比表面积为414m2/g,磁化强度为9emu/g。该材料在25℃条件下对水中三氯生(10ppm)的吸附量达到115mg/g,将100mg已经吸附三氯生饱和的磁性高分子材料外加磁场分离后加入到1ml甲醇溶剂中,震荡后即可完全释放被吸附的三氯生,因此,该材料还可用于三氯生的控制释放。
实施例5
本实施例中制备磁性高分子吸附材料的步骤如下:
(1)、制备磁性fe3o4@油酸纳米粒子:在500ml的烧瓶中加入3g氯化亚铁和1g氯化铁、200ml水,电动搅拌混合均匀,通氮气保护,然后在80℃温度下加入2mol/l的氢氧化钠溶液20ml、油酸3g,所述的油酸的质量为磁性fe3o4纳米粒子的4.5倍;反应7小时后将溶液ph值调为2。将产物用水和甲醇洗涤5次、干燥后即可得到磁性fe3o4@油酸纳米粒子。
(2)、配制油相溶液:将2g磁性fe3o4@油酸纳米粒子溶解于10ml环己醇溶液中,分别加入3.5gn-乙烯吡咯烷酮、2.5g二乙烯苯、0.006gaibn,冰浴混合均匀得到油相溶液。所述的n-乙烯吡咯烷酮质量占n-乙烯吡咯烷酮和二乙烯苯总质量的58%,引发剂bpo占n-乙烯吡咯烷酮和二乙烯苯总质量的0.1%。
(3)、合成磁性高分子吸附材料:在50ml水溶液中加入0.3g乳化剂sds、0.15g分散剂hec;在55℃下加40%上述油相溶液油相溶液,在95℃下加入其余上述油相溶液,所述的乳化剂质量占n-乙烯吡咯烷酮和二乙烯苯总质量的5%,分散剂质量占n-乙烯吡咯烷酮和二乙烯苯总质量的2.5%;100rpm、95℃搅拌80小时后,将反应后溶液进行沉淀、过滤,甲醇洗涤5次,85℃下干燥12小时,最终得到磁性高分子吸附材料。
本实施例中所得磁性高分子吸附材料的粒径集中分布在100μm,比表面积为225m2/g,磁化强度为9.5emu/g。该材料在25℃条件下对水中三氯生(10ppm)的吸附量达到98.5mg/g。
实施例6
本实施例中制备磁性高分子吸附材料的步骤如下:
(1)、制备磁性fe3o4@油酸纳米粒子:在250ml的烧瓶中加入1g氯化亚铁和2.5g氯化铁、150ml水,电动搅拌混合均匀,通氮气保护,然后在80℃温度下加入饱和氨水溶液20ml、油酸10g,所述的油酸的质量为磁性fe3o4纳米粒子的5倍;反应5小时后将溶液ph值调为1。将产物用水和甲醇洗涤5次、干燥后即可得到磁性fe3o4@油酸纳米粒子。
(2)、配制油相溶液:将2g磁性fe3o4@油酸纳米粒子溶解于30mldmf液中,分别加入0.5gn-乙烯吡咯烷酮、4.5g二乙烯苯、0.1gbpo,冰浴混合均匀得到油相溶液。所述的n-乙烯吡咯烷酮质量占n-乙烯吡咯烷酮和二乙烯苯总质量的10%,引发剂bpo占n-乙烯吡咯烷酮和二乙烯苯总质量的2%。
(3)、合成磁性高分子吸附材料:在200ml水溶液中加入0.3g乳化剂sds,在55℃下加45%上述油相溶液油相溶液,在60℃下加入其余上述油相溶液,所述的乳化剂质量占n-乙烯吡咯烷酮和二乙烯苯总质量的6%;60℃、700rpm搅拌48小时后,将反应液进行沉淀、过滤,甲醇洗涤5次,75℃下干燥12小时,最终得到磁性高分子吸附材料。
本实施例中所得磁性高分子吸附材料的粒径集中分布在40μm,比表面积为624m2/g,磁化强度为11.2emu/g。该材料在25℃条件下对水中锌离子的吸附量达到75.2mg/g。
实施例7
本实施例中制备磁性高分子吸附材料的步骤如下:
(1)、制备磁性fe3o4@油酸纳米粒子:在500ml的烧瓶中加入5g氯化亚铁和7g氯化铁、300ml水,电动搅拌混合均匀,通氮气保护,然后在70℃温度下加入2mol/l的氢氧化钠溶液50ml、油酸2g,所述的油酸的质量为磁性fe3o4纳米粒子的0.5倍;反应6小时后将溶液ph值调为2。将产物用水和甲醇洗涤5次、干燥后即可得到磁性fe3o4@油酸纳米粒子。
(2)、配制油相溶液:将1g磁性fe3o4@油酸纳米粒子溶解于20ml甲苯溶液中,分别加入4.5gn-乙烯吡咯烷酮、0.5g二乙烯苯、0.1gaibn,冰浴混合均匀得到油相溶液。所述的n-乙烯吡咯烷酮质量占n-乙烯吡咯烷酮和二乙烯苯总质量的90%,引发剂aibn占n-乙烯吡咯烷酮和二乙烯苯总质量的2%。
(3)、合成磁性高分子吸附材料:在200ml水溶液中加入1g乳化剂htab;在35℃下加三分之一上述油相溶液油相溶液,在75℃下加入其余上述油相溶液,所述的乳化剂的质量占n-乙烯吡咯烷酮和二乙烯苯总质量的20%;75℃、1500rpm搅拌36小时后,将反应后溶液进行沉淀、过滤,甲醇洗涤5次,75℃下干燥24小时,最终得到磁性高分子吸附材料。
本实施例中所得磁性高分子吸附材料的粒径集中分布在50μm,比表面积为772m2/g,磁化强度为19.5emu/g。
实施例8
本实施例中制备磁性高分子吸附材料的步骤如下:
(1)、制备磁性fe3o4@油酸纳米粒子:在500ml的烧瓶中加入5g氯化亚铁和8g氯化铁、200ml水,电动搅拌混合均匀,通氮气保护,然后在80℃温度下加入4mol/l的氢氧化钠溶液20ml、油酸9g,所述的油酸的质量为磁性fe3o4纳米粒子的1.45倍;反应12小时后将溶液ph值调为1。将产物用水和乙醇洗涤5次、干燥后即可得到磁性fe3o4@油酸纳米粒子。
(2)、配制油相溶液:将2g磁性fe3o4@油酸纳米粒子溶解于30mldmso溶液中,分别加入2gn-乙烯吡咯烷酮、8g二乙烯苯、0.5gbpo,冰浴混合均匀得到油相溶液。所述的n-乙烯吡咯烷酮质量占n-乙烯吡咯烷酮和二乙烯苯总质量的20%,引发剂bpo占n-乙烯吡咯烷酮和二乙烯苯总质量的5%。
(3)、合成磁性高分子吸附材料:在150ml水溶液中加入0.3g乳化剂sds、0.22g分散剂peg、0.075g分散剂hec;在45℃下加45%上述油相溶液油相溶液,在65℃下加入其余上述油相溶液,所述的乳化剂质量占n-乙烯吡咯烷酮和二乙烯苯总质量的3%,分散剂质量占n-乙烯吡咯烷酮和二乙烯苯总质量的3%;600rpm、65℃搅拌36小时后,将反应后溶液进行沉淀、过滤,甲醇洗涤5次,65℃下干燥12小时,最终得到磁性高分子吸附材料。
本实施例中所得磁性高分子吸附材料的粒径集中分布在2μm,比表面积为950m2/g,磁化强度为15emu/g。该材料在25℃条件下对水中苯酚(100ppm)的吸附量达到380.5mg/g。
实施例9
本实施例中制备磁性高分子吸附材料的步骤如下:
(1)、制备磁性fe3o4@柠檬酸纳米粒子:在500ml的烧瓶中加入2.5g氯化亚铁和4g氯化铁、200ml水,电动搅拌混合均匀,通氮气保护,然后在80℃温度下加入4mol/l的氢氧化钠溶液20ml、柠檬酸钠5g,所述柠檬酸的质量为磁性fe3o4纳米粒子的1.5倍;反应10小时后将溶液ph值调为2。将产物用水和乙醇洗涤5次、干燥后即可得到磁性fe3o4@柠檬酸纳米粒子。
(2)、配制油相溶液:将2.2g磁性fe3o4@柠檬酸纳米粒子溶解于30mldmso溶液中,分别加入2gn-乙烯吡咯烷酮、8g二乙烯苯、0.3gbpo,冰浴混合均匀得到油相溶液。所述的n-乙烯吡咯烷酮质量占n-乙烯吡咯烷酮和二乙烯苯总质量的20%,引发剂bpo占n-乙烯吡咯烷酮和二乙烯苯总质量的3%。
(3)、合成磁性高分子吸附材料:在150ml水溶液中加入0.3g乳化剂sds、0.22g分散剂peg、0.075g分散剂hec;在45℃下加45%上述油相溶液油相溶液,在65℃下加入其余上述油相溶液,所述的乳化剂质量占n-乙烯吡咯烷酮和二乙烯苯总质量的3%,分散剂质量的占n-乙烯吡咯烷酮和二乙烯苯总质量的3%;65℃、600rpm搅拌36小时后,将反应后溶液进行沉淀、过滤,甲醇洗涤5次,65℃下干燥12小时,最终得到磁性高分子吸附材料。
本实施例中所得磁性高分子吸附材料的粒径集中分布在20μm,比表面积为345m2/g,磁化强度为10emu/g。该材料在25℃条件下对水中苯酚(100ppm)的吸附量达到220.5mg/g。
表1为实施例1~9制得材料的参数及性能。
表1实施例1~9制得材料的参数及性能
由于该材料具有对多种物质的吸附和控制释放性能,因此可以用于不同物质的分离。
以上示意性的对本发明及其实施方式进行了描述,该描述没有限制性,附图中所示的也只是本发明的实施方式之一,实际的流程并不局限于此。所以,如果本领域的普通技术人员受其启示,在不脱离本发明创造宗旨的情况下,不经创造性的设计出与该技术方案相似的结构方式及实施例,均应属于本发明的保护范围。
-
 0访客 来自[中国] 2021年07月09日 08:06这种树脂的配方,工艺我都有,源自澳大利亚。谁有想法投资或者技术转让联系我。
0访客 来自[中国] 2021年07月09日 08:06这种树脂的配方,工艺我都有,源自澳大利亚。谁有想法投资或者技术转让联系我。