水热制备基于钼铋钴铁的混合氧化物催化剂的方法与流程
多元素氧化物催化剂、尤其是基于氧化钼和氧化铋的催化剂被用于通过丙烯、和异丁烯(2-甲基-1-丙烯)或叔丁醇(2-甲基-2-丙醇)的分别部分氧化来工业制备丙烯醛和丙烯酸、或甲基丙烯醛和甲基丙烯酸,以及通过丙烯的氨氧化来工业制备丙烯腈。自1959年俄亥俄标准油公司(thestandardoilofohiocompany)(简称sohio)开发用于这些反应的钼酸铋催化剂以来,这些混合氧化物或多元素氧化物催化剂已获得极大关注,且因此对其催化性质已被详细研究。这些研究涉及除铋和钼之外的其它元素对丙烯醛或丙烯腈形成的产率和选择率,以及所讨论的催化剂在反应中的稳定性的影响。这些研究的一个结果是,不管另外的元素如何,这些多元素氧化物催化剂的表面层始终基于铋和钼的氧化物。因此,这些元素被认为是这些催化剂的活性元素。
通常,借助于所谓的共沉淀来制备多元素氧化物催化剂,其中将待制备的催化剂的元素的前体化合物溶解在水中,然后从该溶液中沉淀出包含这些元素的催化活性组合物,然后干燥并煅烧该活性组合物。另外,尽管程度小得多,多元素氧化物催化剂还通过固态合成、溶胶-凝胶合成和喷雾干燥从包含待制备的多元素氧化物催化剂的前体化合物的溶液来制备。从这些合成获得的多元素氧化物催化剂应该具有最大表面积,以便能够确保催化反应中的高活性。通常,多元素氧化物催化剂借助于催化剂呈结晶材料的形式来实现高活性。然而,为了借助于上述合成提供结晶材料,无论是能够将起始化合物转化为期望的多元素氧化物,例如将硝酸盐完全转化为相应的氧化物,还是为了催化剂的最终煅烧,都始终需要高温,尤其是大于400℃的温度。然而,这样高的温度导致所得混合氧化物相的催化性能退化。相信该作用可归因于催化剂表面中铋的富集。
美国专利4,418,007a公开了包含钼和/或钨的氧化物的混合氧化物催化剂的制备,其中在第一阶段中制备具有所有金属的盐以包含活性催化剂的水悬浮液,并且通过添加氨将该溶液的ph调节到6至8的值,之后过滤所述悬浮液以获得期望活性催化剂的糊料,并且在第二阶段中干燥所述糊料并煅烧至少一次。
美国专利4,166,808a公开了用于制备具有对应于式mo12co10fe1bi1ox的活性相的催化剂的方法,其中在第一步骤中提供金属钼、钴、铁和铋的前体化合物的溶液,所述溶液具有1.1的ph。将所述溶液加热至80℃,以蒸发水,获得非液体糊料,将其进一步加热,获得固体,随后将该固体煅烧并进一步加工,得到负载型催化剂。
美国专利5,245,083a公开了混合氧化物催化剂的制备,该催化剂通过两种组合物的组合获得,这两种组合物是通过从每种不同金属盐的水溶液共沉淀、之后煅烧制备的。所述混合氧化物催化剂是通过以下方式获得的:将两种组合物以特定比率且与一定量的水混合,从而获得悬浮液,然后将其蒸发至干燥并煅烧,得到最终催化剂。
同样,已公开的美国专利申请2009030230a公开了,通过从含有也将在最终催化剂中的所有金属的盐的水溶液中共沉淀固体,来制备混合氧化物催化剂。
借助于共沉淀制备的标准多元素氧化物催化剂的又一问题是它们受时间限制的使用时段。催化剂在这一受时间限制的使用时段内老化,意味着它们不再具有在特定时间与在催化反应中开始使用时相同的催化活性。在这种情况下,不得不将老化或废弃的固定催化剂床更换为新鲜催化剂。
观察到具有含氧化态钼的活性组合物的混合氧化物或多元素氧化物催化剂的老化,尤其是当该催化剂用于存在或形成蒸汽的部分气相氧化时。在多数情况下,蒸汽是在部分气相氧化中形成的。相信部分气相氧化中存在或形成的蒸汽形成挥发性的moo2(oh)2,其中钼以氧化形式存在于所述催化剂的活性组合物中。因此,专业文献认为,在非均相催化气相氧化的长期操作期间,钼作为moo2(oh)2从多元素氧化物催化剂的活性物质中不断升华(参见导致钼从钼酸铋催化剂中损失的机理和动力学的研究(investigationsofthemechanismandkineticsleadingtoalossofmolybdenumfrombismuthmolybdatecatalysts),l.zhang等,catalysisa:general(1994),117,163-171)。混合氧化物催化剂中活性钼组分的损失最大,尤其是在所谓的固定催化剂床的热点(即在反应气体混合物的流动方向上在固定催化剂床上进行的反应的热释放最大的点)的区域。固定催化剂床可具有多个热点。在通过这些区域后,固定催化剂床和其在气体流动方向上的环境的温度也显著下降,使得氧化钼在反应管内壁上的较冷点处再次沉淀为薄涂层或膜(参见ep2155376a1)。在固定催化剂床的一个区域已经历钼从活性组分中渗出后,热点(hotspot)沿着固定催化剂床进一步迁移至固定催化剂床在气体流动方向上的温度先前较低且因此尚未经历钼渗出的区域中。这些区域或区段也逐渐缺乏钼。由于催化剂中活性钼组分的逐区缺乏/流失,该过程也被称为区段老化(带老化)。
因此,随着多元素氧化物催化剂操作时间的增加,不仅所述催化剂中钼组分的比例降低,而且所述催化剂关于待催化的反应的活性也降低。这表现为关于待氧化组分(例如丙烯)的转化率降低。转化率的下降可通过增加含有催化剂的反应器管周围的盐浴温度来抵消。在这种情况下,使特定转化所需盐浴的温度增加至一个值,该值在假定其它方面不变的反应条件下,实现在单次通过反应器时与催化剂活性降低之前相同的待氧化化合物的转化率。然而,增加盐浴温度以抵消含有氧化形式的钼的混合氧化物催化剂的活性组合物的钝化对所讨论的混合氧化物催化剂的寿命起反作用。这是因为固定催化剂床中较高的温度加速了催化剂的老化或钝化,这过早地使得需要用新鲜催化剂完全或至少部分替换老化的固定催化剂床。这不可避免地导致固定催化剂床更频繁的变化和更频繁的生产停工。除了相关的生产损失之外,更频繁地处置废催化剂和更频繁地生产新催化剂也构成相当大的经济负担。
作为标准制备方法的替代,还可借助于水热合成制备基于钼酸铋的混合氧化物催化剂。与上述制备方法相比,水热合成中的反应条件通常不太严格。因此,水热合成能够实现对具有高纯度、受控形态和高结晶度的材料具有良好可再现性的实际可用途径。然而,迄今为止,仅钼酸铋,即仅二元体系,已借助于水热合成制备。即使利用钼酸铋实现对丙烯的催化部分气相氧化中丙烯醛形成的良好选择率,这些体系仍然不适合以工业规模使用。这是因为在该反应中用钼酸铋实现的丙烯转化率不大于约15%。然而,在这样低的丙烯转化率的情况下,则有必要使特别大量的未转化丙烯再循环。这导致能量消耗和资金成本两者的大幅上升,意味着工业规模上的实施意义不大。
此外,工业上用于丙烯氧化的混合氧化物催化剂还含有除钼和铋之外的元素。如果混合氧化物催化剂的水热合成从二元体系如钼酸铋变换为含有钴和铁以及铋和钼的更复杂的多组分体系,则这对所形成的相、掺入这些相中的催化活性元素的量以及所述相的表面积的大小具有直接作用。
由于在活性和选择率方面优化的多元素氧化物催化剂被认为是提高的表面积、适合的晶体相和催化活性元素的关键量的组合,因此需要用于提供具有高结晶度的相应多元素氧化物催化剂的方法。
该问题根据本发明,通过用存在于待制备的多元素氧化物催化剂中的金属的至少四种金属前体化合物、尤其是至少四种过渡金属前体化合物的水溶液和/或水悬浮液进行水热合成来解决,所述水溶液和/或水悬浮液的ph已被调节到约6至约8的值。
因此,本发明提供了用于制备通式(i)的多元素氧化物催化剂的方法
moabibcocfedniexfx’gx”hx”’ix””jox(i)其中
x=w和/或p,
x’=li、na、k、rb、cs、mg、ca、sr和/或ba,
x”=ce、mn、cr和/或v,
x”’=nb、se、te、sm、gd、la、y、pd、pt、ru、ag和/或au,
x””=si、al、ti和/或zr,
并且
a=12,
b=1至4,
c=4至10,
d=1至4,
e=0至4,
f=0至5,
g=0至2,
h=0至5,
i=0至2,
j=0至800,
并且
x=通过除氧之外的元素的化合价和频率确定的数,包括以下步骤
a)以实现根据所述通式(i)的化学计量所需的量,提供存在于所述待制备的多元素氧化物催化剂中的元素的前体化合物的水溶液和/或水悬浮液的混合物,
b)将从步骤a)获得的所述混合物的ph设定为6±0.5至8±0.5的值,
c)在高压釜中,在大于100℃至600℃的温度下,在溶剂热反应条件下,使所述步骤b)的含有前体化合物的混合物反应,以形成所述通式(i)的多元素氧化物催化剂,和
d)从所述水溶液和/或悬浮液中分离所述多元素氧化物催化剂。
在本发明的上下文中,术语“多元素氧化物催化剂”和“混合氧化物催化剂”同义使用,并且是指至少由根据本发明的元素钼、铋、钴和铁组成的催化剂。
因此,根据本发明的方法优选用于制备通式(i)biamobcocfedox的多元素氧化物催化剂,其中a=1至4,b=12,c=4至10且d=1至4。
更具体地,根据本发明的方法提供具有如下限定的通式(i)biamobcocfedox的多元素氧化物催化剂:a=1至2,b=12,c=5至8且d=2至3。
在本发明的上下文中,根据本领域技术人员的常识使用术语“水热合成”,并且是指在水中在高于100℃的温度和高于1巴的压力下进行的非均相反应。这些反应条件,即高于100℃和高于1巴的温度和压力,还被称为溶剂热或水热反应条件。在水热合成的情况下,通常需要使用高压釜。它们用于保护反应容器,高压釜本身经常是反应容器。反应容器中的压力通过温度来调控——当使用反应介质时,与外部隔离的反应容器中200℃的温度已经导致800巴或更高的表压。使用高压釜本身作为耐压反应容器使得能够在宽的压力和/或温度范围内进行水热合成。此外,高压釜相对于与外部隔离的其它反应容器(例如安瓿)具有以下优点:还可以在反应期间连续向反应介质中供应另外易溶解的组分,例如酸、碱或其它络合物形成物质,所述组分还被称为矿化剂。因此,根据本发明的方法或根据本发明的方法的至少步骤c)在高压釜中进行。
关于存在于待制备的多元素氧化物催化剂中的元素的前体化合物,根据本发明的方法原则上不受任何限制。有利地,所用的前体化合物是那些可在水热合成后的任何热处理中无残余地去除阴离子的化合物。因此,优选地,将盐用作存在于待在本发明的方法的步骤a)中制备的多元素氧化物催化剂中的元素的前体化合物。特别地,将硝酸盐、碳酸盐、甲酸盐、草酸盐或类似化合物用作根据本发明的方法中的前体化合物。
因此,在根据本发明的方法的实施方案中,将盐用作步骤a)中的前体化合物。
在本发明的上下文中,表述“6±0.5至8±0.5的ph”涵盖5.5至8.5(包含端值)的所有ph(其可由整数和/或实数表示),尤其是5.5、6、6.5、7、7.5、8和8.5的ph。
已借助于水热合成在这些ph下制备的多元素催化剂以至少50%至最高约82%的选择率将丙烯催化氧化为丙烯醛。
优选地,将根据本发明的方法的步骤b)中的ph调节到6至8的值,即满足条件6≤ph≤8的值,尤其是调节至大于6且小于8的值。
通过在甚至更窄的ph范围内,尤其是在7±0.5的ph下进行根据本发明的方法的步骤b),不仅实现了对于丙烯醛形成具有至少65%至高达约82%的值的选择率的催化剂,而且也将丙烯转化率增加至高达约65%的值。
因此,在根据本发明的方法的一个实施方案中,在步骤b)中建立7±0.5的ph。
在本发明的上下文中,已发现合成中ph的选择显著影响催化剂的组成。水热合成中上升的ph倾向于导致所制备的催化剂中催化活性钼组分的含量明显降低;同时,观察到铋组分的比例明显增加。专业文献通常认为钼的作用是用于气相部分氧化的催化活性组分。因此,应该预计到在ph=5下制备的具有最高钼含量的催化剂也应该在丙烯的部分气相氧化中具有最佳的催化性质。然而,研究已显示,在7±0.5的ph下制备的根据本发明的催化剂同时具有最高的丙烯醛选择率和最高的丙烯转化率。
此外,已发现,在根据本发明方法的步骤b)中建立的ph还影响催化剂的比表面积:随着ph的上升,催化剂的比表面积也增加。催化剂的高表面积通常与高催化活性相关。因此,应该预计到在更高ph(ph=8或更高)下制备的催化剂应该在部分气相氧化中展现出最高的丙烯转化率。然而,已在研究中发现,令人惊讶地,在ph7±0.5下制备的催化剂展现出在丙烯的部分气相氧化中最高的丙烯转化率。
此外,已发现,在ph=7下制备的多元素氧化物催化剂的催化反应中的放热性低于通过共沉淀制备的比较催化剂的情况。因此,当用于烯烃的部分气相氧化中时,使相应的催化剂经受较低的热应力。结果,催化剂例如不会那样快地耗尽催化活性的钼组分。总之,较低的热应力对催化剂的寿命具有积极作用。
因此,7±0.5的ph构成了对水热合成中ph的特别有利的选择。将ph设定为7±0.5似乎产生具有钼含量和比表面积的特别好的组合的催化剂,因为在这些条件下获得的催化剂产生特别好的丙烯醛选择率,且同时也产生特别好的丙烯转化率。
可能的情况是,存在于待制备的催化剂中的元素的前体化合物在作为溶剂热反应介质的纯水中未获得所需的溶解度。因此,为了形成具有比纯水更好溶解性的所讨论元素的其它络合物,优选将酸、碱或其它络合物形成物质添加至前体化合物的溶液和/或悬浮液中。关于待添加的酸、碱或络合物形成物质,根据本发明的方法原则上不受任何限制。有利地,选择待添加的酸、碱或络合物形成物质,使得它可在水热合成后以低复杂程度被无残余地去除,例如通过简单洗涤或在升高的干燥温度下分解。因此,用于水热合成中的助剂(还被称为矿化剂)是例如硝酸、乙酸、甲酸或类似化合物。优选地,在根据本发明的方法的步骤a)中,添加硝酸以改善前体化合物的溶解度。
除了上述酸之外,水合形式的一些前体化合物和/或前体化合物的一些金属阳离子在水性介质中表现为酸,在一些情况下表现为强酸。通过添加适合的碱,在根据本发明的方法的步骤b)中建立期望的ph。关于这种碱的选择,本发明的方法原则上不受任何限制,条件是确保通过其添加可以建立6±0.5至8±0.5的ph,优选大于6且小于8的ph,且尤其是7±0.5的ph,并且添加的碱不会破坏多元素氧化物催化剂的形成。优选地,在根据本发明的方法中,通过使用碱来建立期望的ph,所述碱可通过洗涤操作和/或通过后续热处理(尤其是在低温下)从所制备的催化剂中容易地去除。优选使用络合碱来建立根据本发明的方法的步骤b)中的ph。不希望受限于特定理论,相信络合碱的存在对所制备的多元素氧化物催化剂的催化性质具有有利作用。因此,优选使用氮碱来建立根据本发明的方法的步骤b)中的ph。优选地,氮碱是氨或伯、仲或叔脂族c1-c4胺,例如甲胺、乙胺、二甲胺、二乙胺、三甲胺或三乙胺。优选使用氨水溶液,尤其是具有至少20%的浓度、尤其是具有20%至30%(例如25%)的浓度的氨水溶液作为碱。
将反应容器、优选高压釜置于烘箱中进行水热合成:可以等温地或在温度梯度下工作。优选在根据本发明的方法的上下文中等温工作,这意味着填充的高压釜连同其内容物一起在密封后被加热直至限定温度,该温度维持特定的时段,然后高压釜连同其内容物一起在特定的时段内被冷却至起始温度,优选室温,或者高压釜连同其内容物一起被冷却至起始温度,优选室温。该程序使得能够在基本上恒定的反应条件下进行根据本发明的方法。
根据本发明,在大于100℃至600℃的温度下进行步骤c)。基于作为溶剂热反应介质的水,在这些温度下进行步骤c)对应于高压釜中大于1巴直至高达约3000巴的内压力。
在根据本发明的方法的实施方案中,在大于100℃至400℃的温度下进行步骤c)。在以水作为溶剂热反应介质进行步骤c)的情况下,该反应温度导致大于1巴至高达约100巴的内压力。
在本发明的上下文中,表述“大于100℃至600℃的温度”和表述“大于100℃至400℃的温度”分别涵盖大于100℃直至高达且包括600℃以及大于100℃直至高达且包括400℃的所有值,这些值可由整数和实数表示。
在本发明的上下文中,表述“大于1巴至3000巴的压力”和表述“大于1巴至100巴的压力”分别涵盖大于1巴直至高达且包括3000巴以及大于1巴直至高达且包括100巴的所有值,这些值可由整数和实数表示。
关于进行步骤c)的时间段,根据本发明的方法不受任何限制,条件是仍然确保该方法得到根据本发明的组合物的混合氧化物催化剂。
因此,在根据本发明的方法的一个实施方案中,步骤c)进行6±0.5小时直至长达48±0.5小时的时期。
有利地,根据本发明的方法之后是对从根据本发明的方法获得的多元素氧化物催化剂进行后处理的另外的步骤。更具体地,这些步骤涉及从水热合成获得的多元素氧化物催化剂的单次或多次(优选多次)洗涤,用于从合成中去除残余物。用于洗涤的适合溶剂原则上是可在一次或多次洗涤操作中从合成中完全去除残余物并使合成的催化剂在其结构和组成方面保持不变的所有那些溶剂。然而,优选那些本身也可以低复杂程度从催化剂中被无残余地去除的溶剂。特别是那些可通过在低温下、尤其是在低于通常用于煅烧的400℃的温度下干燥去除的溶剂优选被用于洗涤。用于洗涤的说明性溶剂是水和丙酮。然后,例如将从合成获得的多元素氧化物催化剂用水洗涤一次或超过一次,优选超过一次,例如三次,然后用丙酮洗涤一次或超过一次,优选洗涤超过一次,例如三次。此后,干燥洗涤的多元素氧化物催化剂。这优选在低于通常用于煅烧的400℃或更高温度的温度下进行。优选地,多元素氧化物催化剂干燥中的温度在20℃至最高390℃范围;干燥温度优选在20℃至350℃范围。低于通常用于煅烧的温度的这些温度已足以使硝酸盐分解。将干燥的持续时间选择为足够长,以确保来自合成的所有残余物和来自洗涤操作的所有溶剂均已从多元素氧化物催化剂中被完全去除。达到这种情况的时刻可通过干燥直至恒重出现来确定,例如借助于热重法。即使在20℃的温度下,长达3天,尤其是12、24、36或48小时的持续时间通常也足以干燥洗涤过的多元素氧化物催化剂。与通过现有合成方法、尤其是共沉淀获得的多元素氧化物催化剂相比,原则上不需要煅烧由根据本发明的方法获得的多元素氧化物催化剂。因此,根据本发明,在比共沉淀温和得多的条件下实现多元素氧化物催化剂的制备。与该标准制备方法相比,在根据本发明的方法中,还避免了铋在催化活性相中的富集。任选地,作为干燥的替代或除了干燥之外,仍然可以在根据本发明的方法中进行煅烧。
因此,在根据本发明的方法的一个实施方案中,所述方法另外包括以下步骤
e)将从步骤d)获得的所述多元素氧化物催化剂洗涤至少一次,和
f)然后干燥和/或煅烧所述多元素氧化物催化剂。
通过根据本发明的方法获得的多元素氧化物催化剂通常为粉末形式。对于工业用途,在添加成形剂和粘合剂后,将粉状催化剂转化为更好管理的形式是有利的。这可通过将催化剂施加至支撑件(例如由氧化铝、氧化锆、二氧化钛或二氧化硅构成),或者通过其它成形步骤如压片或挤出来实现。支撑件的几何形状在这里不受限制,而是取决于反应器的规格,例如管径、催化剂床的长度。因此,支撑件可以是例如锥体、圆柱体、鞍形件、球或多面体,但其还可以是反应空间的壁,例如呈壁反应器的形式。
所用的粘合剂可以是各种油、纤维素、聚乙烯醇、糖、丙烯酸酯、其烷基衍生物、其缩合物或其混合物。优选的粘合剂是丙烯酸酯、聚乙烯醇、纤维素、其烷基衍生物和其混合物。
例如,通过将催化剂在适合的溶剂或溶剂混合物中的悬浮液与粘合剂一起喷雾至支撑体上,或者通过将催化剂悬浮液喷雾至被粘合剂润湿的支撑体上,来制备负载型催化剂。适合的溶剂是例如水、甲醇、乙醇或类似的化合物或其混合物。例如,通过将催化剂与上述粘合剂混合并随后成形来制备非负载型催化剂。
随后对通过各自的成形操作获得的催化剂进行干燥和/或煅烧,以完成溶剂且尤其是粘合剂的去除。
在又一实施方案中,根据本发明的方法因此包括以下另外的步骤:
i)将从步骤d)、e)或f)获得的所述多元素氧化物催化剂施加至载体上以获得负载型催化剂,
或
ii)形成从步骤d)、e)或f)获得的所述多元素氧化物催化剂,以获得非负载型催化剂,
和
iii)干燥和/或煅烧从步骤i)或ii)获得的所述多元素氧化物催化剂。
在具有能量色散x射线光谱的扫描电子显微镜(sem-edx)中对通过根据本发明的方法获得的多元素氧化物催化剂的研究已显示,所述催化剂具有被认为是催化活性元素的钼和铋两者与铁一起同时存在的区域。相信根据本发明的催化剂在烯烃的部分气相氧化中的改进的催化性质可归因于元素钼、铋和铁在催化剂区域中的联合存在。
因此,本发明还进一步提供了以下通式的多元素氧化物催化剂
moabibcocfedniexfx’gx”hx”’ix””jox(i)
其中
x=w和/或p,
x’=li、k、na、rb、cs、mg、ca、ba和/或sr,
x”=ce、mn、cr和/或v,
x”’=nb、se、te、sm、gd、la、y、pd、pt、ru、ag和/或au,
x””=si、al、ti和/或zr,
并且
a=12,
b=1至4,
c=4至10,
d=1至4,
e=0至4,
f=0至5,
g=0至2,
h=0至5,
i=0至2,
j=0至800,
并且
x=通过除氧之外的元素的化合价和频率确定的数,
其特征在于,它包含钼、铋和铁同时存在的区域,所述区域具有10nm至25μm的直径。
相比之下,还含有所述三种元素的根据现有技术的普通多元素氧化物催化剂不具有所有三种元素同时存在的任何区域。相反,普通的多元素氧化物催化剂具有仅存在一种元素的区域。
在根据本发明的多元素氧化物催化剂的一个实施方案中,钼、铋和铁同时存在的区域具有1μm至22μm的直径
在一个实施方案中,通过根据本发明的方法获得和/或可获得本发明的多元素氧化物催化剂。
在这种情况下,当催化剂的水热合成中的ph为7±0.5时,元素钼、铋和铁尤其一起存在于根据本发明的催化剂的区域或区段中。在这种情况下,在根据本发明的方法的步骤b)中建立7±0.5的ph。
在一个实施方案中,通过根据本发明的方法获得和/或可获得根据本发明的多元素氧化物催化剂,其中在根据本发明的方法的步骤b)中建立7±0.5的ph。
在根据本发明的多元素氧化物催化剂的一个实施方案中,e至i各自为0。
在根据本发明的多元素氧化物催化剂的又一实施方案中,a=12,b=1至2,c=5至8且d=2至3。
与通过标准制备方法获得的多元素氧化物催化剂、尤其是通过共沉淀获得的多元素氧化物催化剂相比,通过根据本发明的方法可获得和/或获得的多元素氧化物催化剂不需要进行煅烧;相反,在比用于煅烧中的温度更低的温度下进行的干燥已经足以使催化剂达到用于催化所需的形式。煅烧和用于催化通常影响所讨论的催化剂的相,使得用于催化的催化剂通常不再与从标准制备方法获得的原始催化剂或催化开始时存在的催化剂相同。现已发现,令人惊讶地,根据本发明的催化剂且尤其是通过根据本发明的方法获得和/或可获得的多元素氧化物催化剂是基本上相稳定的。这意味着这些多元素氧化物催化剂在催化反应期间经历低程度的相变,如果有的话。低程度的相变在于催化反应期间875cm-1(femoo4)处拉曼信号的强度增益。相比之下,通过共沉淀制备方法获得的比较催化剂,例如通过wo2007/042369a1的方法获得的催化剂mo12bi1.5co5fe1.8ox,在催化反应期间经历显著的相重组:moo3在995cm-1处的拉曼信号消失,bi2mo3o12在814cm-1和900cm-1处的拉曼信号减弱,并且femoo4在875cm-1处的拉曼信号出现。
在一个实施方案中,根据本发明的多元素氧化物催化剂、尤其是通过根据本发明的方法获得和/或可获得的多元素氧化物催化剂因此是基本上相稳定的,尤其是在用于烯烃或叔丁醇的部分气相氧化期间。
现有技术的多元素氧化物催化剂、特别是通过共沉淀制备的那些含有moo3相。所述相在它们用于部分气相氧化或氨氧化中之前或之后存在于共沉积催化剂中。相比之下,根据本发明的催化剂、尤其是通过根据本发明的方法获得和/或可获得的催化剂,在它们用于部分气相氧化或氨氧化反应之前以及在它们用于部分气相氧化或氨氧化反应之后不含moo3相。然而,根据本发明的催化剂和通过根据本发明的方法获得和/或可获得的催化剂含有α-钼酸铋相和β-钼酸钴相。
在实施方案中,根据本发明的多元素氧化物催化剂、尤其是通过根据本发明的方法获得和/或可获得的多元素氧化物催化剂不含moo3相。
在一个实施方案中,根据本发明的多元素氧化物催化剂、尤其是通过根据本发明的方法获得和/或可获得的多元素氧化物催化剂具有α-钼酸铋相和β-钼酸钴相。
α-钼酸铋相和β-钼酸钴相尤其由通过根据本发明的方法获得和/或可获得的多元素氧化物催化剂具有,所述催化剂已在根据本发明的方法的步骤b)中在7±0.5的ph下获得。
通过根据本发明的方法获得和/或可获得的多元素氧化物催化剂和根据本发明的多元素氧化物催化剂适于烯烃或叔丁醇的部分气相氧化和氨氧化。
在本发明的上下文中,术语“部分氧化”、“部分气相氧化”和“气相部分氧化”以如下方式使用,即它们是指有机化合物在氧的反应作用下的转化,其中待氧化的化合物在反应结束后含有比反应前多至少一个的化学键合的氧原子。与完全氧化相比,部分氧化、部分气相氧化或气相部分氧化不会将存在于待氧化化合物中的所有碳原子都氧化成二氧化碳。
因此,本发明还进一步提供了通过根据本发明的方法获得和/或可获得的多元素氧化物催化剂和/或根据本发明的多元素氧化物催化剂在烯烃或叔丁醇的部分气相氧化和/或氨氧化中的用途。
烯烃的部分气相氧化产生不饱和醛和相应的不饱和羧酸,并且烯烃的氨氧化产生不饱和腈。最经济重要的不饱和醛、羧酸和腈是c3和c4化合物。因此,在本发明的上下文中,通过根据本发明的方法获得和/或可获得的混合氧化物催化剂和/或根据本发明的混合氧化物催化剂被用于通过丙烯的催化气相部分氧化来工业制备丙烯醛和丙烯酸,通过异丁烯(2-甲基-1-丙烯)或叔丁醇(2-甲基-2-丙醇)的催化气相部分氧化来工业制备甲基丙烯醛和甲基丙烯酸,以及通过丙烯的氨氧化来工业制备丙烯腈。以用空气或氧气对丙烯、或异丁烯或叔丁醇的非均相催化氧化的形式,或者以用氨和空气或氧气对丙烯的非均相催化氨氧化的形式,在主要包含基于氧化钼和氧化铋的混合氧化物催化剂的固定催化剂床上实现这些化合物的制备。
因此,在根据本发明的用途的一个实施方案中,烯烃是丙烯和/或异丁烯。
在本发明的上下文中,已发现通过根据本发明的方法获得和/或可获得的多元素氧化物催化剂和/或根据本发明的多元素氧化物催化剂对于不饱和醛的形成的选择率远远高于对于不饱和羧酸的形成的选择率。
因此,优选地,根据本发明的用途是烯烃被部分气相氧化成不饱和醛。更具体地,根据本发明的用途是丙烯被部分气相氧化成丙烯醛和/或异丁烯和/或叔丁醇被部分气相氧化成甲基丙烯醛。
通过以下项目进一步描述本发明:
1.用于制备通式(i)的多元素氧化物催化剂的方法
moabibcocfedniexfx’gx”hx”’ix'““jox(i)其中
x=w和/或p,
x’=li、k、na、rb、cs、mg、ca、ba和/或sr,
x”=ce、mn、cr和/或v,
x”’=nb、se、te、sm、gd、la、y、pd、pt、ru、ag和/或au,
x””=si、al、ti和/或zr,
并且
a=12,
b=1至4,
c=4至10,
d=1至4,
e=0至4,
f=0至5,
g=0至2,
h=0至5,
i=0至2,
j=0至800,
并且
x=通过除氧之外的元素的化合价和频率确定的数,包括以下步骤
a)以实现根据所述通式(i)的化学计量所需的量提供存在于所述待制备的多元素氧化物催化剂中的元素的前体化合物的水溶液和/或水悬浮液的混合物,
b)将从步骤a)获得的所述混合物的ph设定为6±0.5至8±0.5的值,
c)在高压釜中,在大于100℃至600℃的温度下,在溶剂热反应条件下,使所述步骤b)的含有前体化合物的混合物反应,以形成所述通式(i)的多元素氧化物催化剂,和
d)从所述水溶液和/或悬浮液中分离所述多元素氧化物催化剂。
2.根据项目1所述的方法,其中将盐用作步骤a)中的前体化合物。
3.根据项目1或2所述的方法,其中在步骤b)中建立7±0.5的ph。
4.根据项目1至3中任一项所述的方法,其中在大于100℃至400℃的温度下进行步骤c)。
5.根据项目1至4中任一项所述的方法,其中步骤c)进行6±0.5小时直至长达48±0.5小时的时期。
6.根据项目1至5中任一项所述的方法,其另外包括以下步骤
e)将从步骤d)获得的所述多元素氧化物催化剂洗涤至少一次,和
f)然后干燥和/或煅烧所述多元素氧化物催化剂。
7.根据项目1至6中任一项所述的方法,其另外包括以下步骤
i)将从步骤d)、e)或f)获得的所述多元素氧化物催化剂施加至载体上以获得负载型催化剂,
或
ii)形成从步骤d)、e)或f)获得的所述多元素氧化物催化剂,以获得非负载型催化剂,
和
ii)干燥和/或煅烧从步骤i)或ii)获得的所述多元素氧化物催化剂。
8.以下通式的多元素氧化物催化剂
moabibcocfedniexfx’gx”hx”’ix””jox(i)
其中
x=w和/或p,
x’=li、k、na、rb、cs、mg、ca、ba和/或sr,
x”=ce、mn、cr和/或v,
x”’=nb、se、te、sm、gd、la、y、pd、pt、ru、ag和/或au,
x””=si、al、ti和/或zr,
并且
a=12,
b=1至4,
c=4至10,
d=1至4,
e=0至4,
f=0至5,
g=0至2,
h=0至5,
i=0至2,
j=0至800,
并且
x=通过除氧之外的元素的化合价和频率确定的数,
其特征在于,它包含钼、铋和铁同时存在的区域,所述区域具有10nm至25μm的直径。
9.根据项目8所述的多元素氧化物催化剂,其通过根据权利要求1至7中任一项所述的方法获得和/或可获得,其中在所述方法的步骤b)中设定7±0.5的ph。
10.根据项目8或9所述的多元素氧化物催化剂,其中e至i各自为0。
11.根据项目8至10中任一项所述的多元素氧化物催化剂,其中a=12,b=1至2,c=5至8且d=2至3。
12.根据项目8至11中任一项所述的多元素氧化物催化剂,其中所述催化剂不含moo3相。
13.根据项目8至12中任一项所述的多元素氧化物催化剂,其中所述多元素氧化物催化剂具有α-钼酸铋相和β-钼酸钴相。
14.通过项目1至7中任一项所述的方法获得和/或可获得的多元素氧化物催化剂和/或根据项目8至13中任一项所述的多元素氧化物催化剂在烯烃或叔丁醇的部分气相氧化和/或氨氧化中的用途。
15.根据项目14所述的用途,其中所述烯烃是丙烯和/或异丁烯。
附图说明:
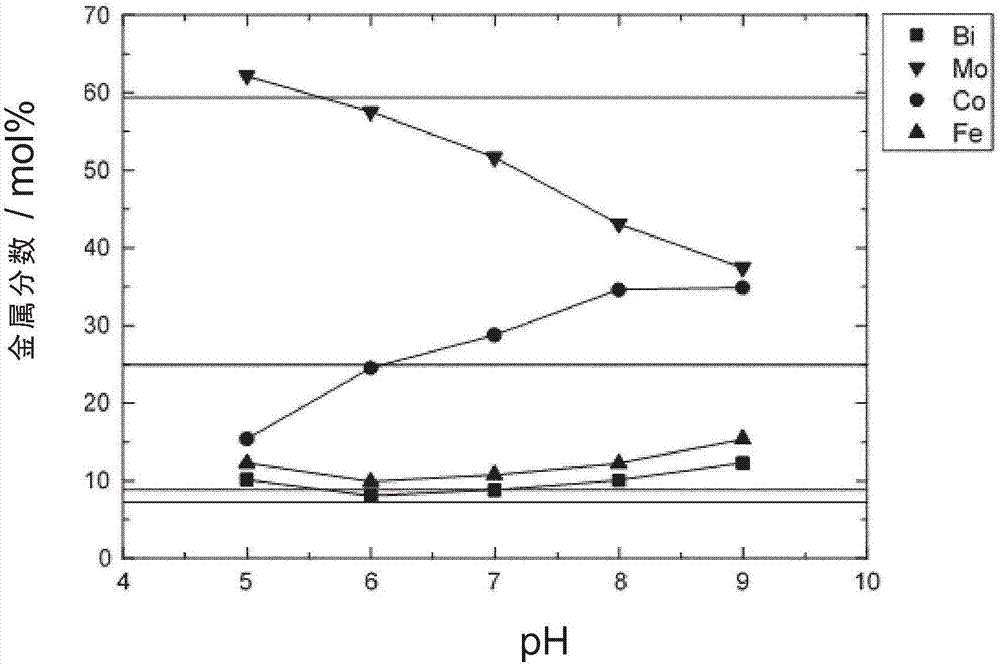
图1:实验1至3和比较实施例c1至c3的催化剂的ph与组成之间的相关性。
图2:实验4至6的催化剂的ph与组成之间的相关性。
图3:水热制备的mo12bi1co8fe3ox催化剂随水热合成中的ph和bet测量前的干燥温度而变化的bet表面积(·=320℃下的干燥;■=150℃下的干燥,其中所述温度是bet测量前的干燥温度)。
图4:来自实验1的多元素催化剂的sem-edx图像(元素铋bi、钼mo和铁fe如图像中所述着色)。
图5:来自实验2的多元素催化剂的sem-edx图像(元素铋bi、钼mo和铁fe如图像中所述着色)。
图6:来自实验3的多元素催化剂的sem-edx图像(元素铋bi、钼mo和铁fe如图像中所述着色)。
图7:实验1至3的在ph=6、7或8下通过水热合成制备的催化剂(在图7中指示为ph=6、ph=7和ph=8)和根据wo2007/042369a1通过共沉淀制备的催化剂(在图7中指示为cp)的相变(在催化反应之前(深色线)和之后的拉曼光谱(浅色线))。
图8:在6至10的ph下水热合成的催化剂mo12bi1.5co5fe3ox的相组成。
图9:通过共沉淀获得的mo12bi1.5co5fe1.8ox催化剂的时间信赖性相变。
图10:在ph=7下水热合成的mo12bi1.5co5fe3ox催化剂的时间依赖性相变。
实施例
实施例1:mo-bi-co-fe氧化物的制备
通过首先将如表中数字所限定的量的五水合硝酸铋(iii)、六水合硝酸钴(ii)和九水合硝酸铁(iii)溶解在20ml硝酸(浓度2m)中并充分搅拌15分钟来制备第一溶液(下文称为溶液i)。平行地,通过将化学计量量的根据表1中的数字的七钼酸铵溶解在20ml脱矿质水中并搅拌15分钟来制备第二溶液(下文称为溶液ii)。在每种情况下,选择铋、钼、钴和铁的前体化合物的摩尔量,使得它们加起来总共为20mmol。将溶液i和溶液ii合并在由
在实施例3中确定了来自实验1至6和比较实验c1至c3的催化剂的组成。
表1:水热合成多元素氧化物催化剂的概述
实施例2:根据本发明的催化剂的表面积的确定
借助于bet分析,确定了在不同ph下通过水热手段制备的mo12bi1co8fe3ox催化剂的比表面积。为此,在150℃和340℃下且在降低的压力下将100至500mg催化剂干燥5小时,其中温度是bet测量之前的干燥温度。利用来利自rubothermgmbh的belsorp-miniii进行bet分析。所用的吹扫气体是氦气。在干燥后,将催化剂样品抽真空并用液氮冷却至77k的温度。逐步增加氮吸附气体的分压,并记录在p/p0=0至1范围内的标准吸附等温线。最后,测量解吸等温线。借助于bet理论在p/p0=0.05至0.3的范围内确定bet表面积。
所确定的bet表面积既与特定催化剂合成中的ph相关,也与干燥温度相关。图3显示这些相关性。
催化剂的比表面积随着合成中建立的ph而上升。在更高温度下进行的干燥同样导致更大的比表面积。
实施例3:所制备的催化剂的元素分析
借助于有电感耦合等离子体的光发射光谱(icp-oes,agilent720/725-es)确定实验1至6和比较实施例c1至c3的催化剂的组成。通过40mhz高频发生器产生等离子体,并且将氩气用作等离子体气体。通过将样品悬浮在6ml浓盐酸、2ml浓硝酸和1ml过氧化氢的混合物中,并随后将所获得的混合物在600瓦的微波中处理45分钟,实现了约40mg用于光发射光谱的样品的消化。
针对各催化剂确定的每摩尔金属分数与针对水热合成建定的ph相关。这些相关性针对实验1至3和比较实验c1至c3的催化剂示于图1中,并且针对实验4至6的催化剂示于图2中。
来自实验1的在ph=6下制备的多元素氧化物催化剂具有大致为mo12bi1.5co5fe1.8ox的组成。从ph=5开始,随着ph的上升,催化钼组分的摩尔比例从约62mol%连续降低至ph=9下的约37mol%。同时,随着ph的上升,钴的摩尔比例从ph=5下的15mol%增加至ph=9下的高达35mol%。随着ph的上升,铁和铋的比例仅增加了数mol%
来自实施例5的在ph=7下制备的多元素氧化物催化剂具有大致为mo12bi1co8fe3ox的组成。从ph=6开始,随着ph的上升,催化钼组分的摩尔比例从55mol%连续降低至ph=8下的约40mol%。同时,随着ph的上升,钴的摩尔比例从25mol%增加至ph=8下的高达约40mol%。铁和铋的比例保持基本上不变,而不管ph分别为约5mol%和约15mol%。
水热合成中的ph与催化剂的特定组成之间的相关性显示,ph对催化剂的组成具有很大影响。特别是催化活性钼组分的摩尔比例受到ph的极大影响。
实施例4:sem-edx分析
在具有能量色散x射线光谱的扫描电子显微镜(sem-edx)中检查来自实验1至3的多元素氧化物催化剂的表面。
已借助于sem-edx制成的实验1至3的多元素氧化物催化剂表面的图像示于图4至6中。在这些图像中,元素钼mo、铋bi和铁fe已被着色。所有三种元素一起存在的催化剂区域——对应于单个元素的颜色的重叠——以白色示于图中。只有在ph=7下制备的来自实验2的多元素氧化物催化剂具有白色区域,即所有三种元素(钼、铋和铁)一起存在的区域(参见图5)。这些区域的直径被确定为在10nm至25μm范围内。
实施例5:相确定
借助于拉曼光谱确定在6、7或8的ph下水热合成并通过共沉淀合成的催化剂mo12bi1.5co5fe3ox(图7)、在6至10的ph下水热合成的实验1至3和比较实验c2至c3的催化剂mo12bi1.5co5fe3ox(图8)、通过共沉淀合成的催化剂mo12bi1.5co5fe1.8ox(图9)以及在ph=7下水热合成的催化剂mo12bi1.5co5fe3ox(图10)的相。为此,使用装配有研究级leicadm2500显微镜的来自renishaw的renishawinvia反射光谱仪系统。利用17mw氦-氖激光器在623nm下分析样品。为此,通常记录50至1300cm-1的光谱范围。
对于图8(在6至10的ph下水热合成的实验1至3和比较实验c2至c3的催化剂mo12bi1.5co5fe3ox)的记录,在室温下用leica50×物镜将激光束聚焦在样品上。在每种情况下,在约300*300μm的区域上记录了超过2000个光谱,并在该区域上取平均值。借助于文献值将获得的拉曼光谱分配给相应的bi、mo、co和fe相。
对于图7(在6、7或8的ph下水热合成的实验1至3的催化剂mo12bi1.5co5fe3ox,)、图9(通过共沉淀合成的催化剂mo12bi1.5co5fe1.8ox)和图10(在7的ph下水热合成的催化剂mo12bi1.5co5fe3ox)中所示的光谱,在每种情况下将30mg具有150至300μm的粒径的催化剂引入直径为2mm的石英玻璃毛细管中。使具有比率为72/18/8/8的n2/o2/c3h6/h2组成的每分钟10ml的气流通过该石英玻璃毛细管。用oxford气体风机(gasblower)将石英玻璃毛细管加热至高达445℃。借助于连接至拉曼光谱仪的来自renishwgmbh的视频光纤,将激光束聚焦至该毛细管上。图7中的光谱各自在催化反应之前(深色线)和之后(浅色线)在80℃下被记录。在借助于气体风机限定的以下温度分布下的催化反应期间记录图9和图10中的光谱:180℃(0min)→4kmin-1→320℃(20min)→4kmin-1→345℃(46min)→4kmin-1→370℃(46min)→4kmin-1→395℃(46min)→4kmin-1→420℃(23min)→4kmin-1→445℃(23min)→-10kmin-1→180℃(18min)→-10kmin-1→60℃。
实施例6:根据本发明的催化剂的测试
在丙烯的部分气相氧化中测试实验1至3的根据本发明的催化剂(hs-bi1.5mo12co5fe3ox-ph6、hs-bi1.5mo12co5fe3ox-ph7和hs-bi1.5mo12co5fe3ox-ph8)、根据wo2007/042369a借助于共沉淀制备的催化剂mo12bi1.5co5fe3ox和借助于水热合成制备的二元催化剂(hs-bi1mo1ox-ph6)。对于每个测试,向内径为6mm的反应器中装入每种情况下800mg筛分粒度级为300至450μm的待测试催化剂,使得其内部基本上完全被催化剂填充。所得固定催化剂床具有2.7cm的高度。固定催化剂床的上端和下端各有一个热电偶,所述热电偶与所述固定催化剂床接触。通过外部供热,将固定催化剂床的温度调节至约380℃的值。在三次实验中,以每分钟100ml、150ml和200ml(stp)的流速,向每个装有特定催化剂的反应器中供应比率为70/14/8/8的组成n2/o2/c3h6/h2的反应气体混合物;基于所用各自催化剂的质量,不同流速的改良停留时间(作为所用催化剂的质量除以流速的商)因此分别为0.48g·s·ml-1、0.32g·s·ml-1和0.24g·s·ml-1。在测试期间,在固定催化剂床温度波动的情况下,通过相应地调节外部热源的温度,使固定催化剂床的温度保持基本恒定在380℃。离开反应器的反应混合物通过加热导管传送至agilent7890b气相色谱仪。
气相色谱仪装配有两个样品回路,所述样品回路用于永久气体,尤其是氮气、氧气、一氧化碳和二氧化碳,以及用于烃,尤其是丙烯、丙烯醛和丙烯酸。第一样品回路由两个串联连接的agilenthayesepq分离柱和agilentmolsieve5a型的最终微填充柱组成。第一hayesepq分离塔用于从烃中去除永久气体;第二hayesepq分离柱用于分离永久气体:二氧化碳是从第二hayesepq分离柱洗脱的第一种气体,然后其它永久气体一起从第二hayesepq分离柱中被洗脱,并通过阀到达用于分离永久气体n2、o2、co的molsieve5a型最终分离柱。利用热导率检测器检测co2和从molsieve5a分离柱洗脱的永久气体。第二个样品回路由agilenthp-ffap毛细管柱组成。利用火焰离子化检测器检测从该分离柱洗脱的化合物(丙烯、丙烯醛和丙烯酸)。用已知浓度的n2、co、co2、丙烯和丙烷的气体混合物以及n2和o2的气体混合物并且用丙烯、丙烯醛和丙烯酸在甲醇中的标准溶液校准气相色谱仪。在该校准之后是在单个催化剂测试中分离和确定反应混合物的单个组分。所用丙烯的量以及检测到的丙烯、丙烯醛、丙烯酸和碳氧化物(co和co2)的量用于确定丙烯转化率以及丙烯醛、丙烯酸和碳氧化物(co和co2)形成的选择率。这些值汇编在下表2至4中,其中hs指示通过水热合成获得,并且cp指示通过共沉淀获得。
表2:在w/f=0.48g·s·ml-1下测试的催化剂催化性质的概述。
表3:在w/f=0.32g·s·ml-1下测试的催化剂催化性质的概述。
表4:在w/f=0.24g·s·ml-1下测试的催化剂催化性质的概述。
- 还没有人留言评论。精彩留言会获得点赞!