本发明属于分离膜改性技术领域,具体涉及基于巯基-炔基的点击化学反应在pvdf膜表面点击碳纳米管,进而制备一种pvdf膜表面点击碳纳米管分离膜的方法。
背景技术:
聚偏氟乙烯(pvdf)因其出色的物理化学稳定性,已经在高性能分离膜材料中被广泛研究及应用;但聚偏氟乙烯分子链的疏水性很大,导致聚偏氟乙烯微孔膜在水处理过程中容易吸附有机物质而使水通量逐渐衰减,进而限制了其在水处理膜领域的应用。
纳米材料是纳米科技发展的重要基础之一。纳米材料主要指几何尺寸达到纳米级别的尺度水平并且具有特殊性能的材料。在诸多无机纳米粒子中,碳纳米管(cnt)具有独特的本征中空结构。由于碳纳米管的原子级光滑内表面,水分子在碳纳米管内腔中能够无摩擦通过,而其它水合离子则需要克服相应能垒后通过。研究结果表明,水分子在碳纳米管内的传输速度可与蛋白生物膜水通道中相当。因此通过碳纳米管改性pvdf膜不仅能够构建水分子通道,提高水通量和选择分离性能,而且利用其电性能可以有效改善pvdf膜的抗污染性能。
由于实验装置简单且适用于多种类型的单体,可控/活性自由基聚合已得到广泛运用。其中,可逆加成-断裂链转移自由基聚合(raft)和原子转移自由基聚合(atrp)占据了领导性的地位。点击化学和活性/可控自由基聚合的结合是一种有效制备功能材料的途径。
我们基于对碳纳米管改性方法的研究探索,采用raft聚合和点击化学相结合的方法在pvdf膜表面接枝亲水性功能化碳纳米管。通过巯基-炔基绿色无催化剂的反应,在无铜(i)催化的条件下,通过光引发剂在紫外光照射下完成反应,该反应具有反应时间短、无催化剂、反应条件适当等优点,通过该反应制备的pvdf膜可以在其表面引入大量的亲水性聚合物基团,结合碳纳米管的水通道作用和亲水性聚合物基团的亲水性能,拓宽了pvdf膜在水处理膜领域的应用。
技术实现要素:
本发明的目的在于克服现有技术中存在的聚偏氟乙烯因分子链的疏水性很大,导致聚偏氟乙烯微孔膜在水处理过程中容易吸附有机物质而使水通量逐渐衰减,进而限制了其在水处理膜领域的应用的技术问题,提供一种pvdf膜表面点击碳纳米管分离膜的制备方法,通过该方法制备的pvdf膜在膜表面接枝覆盖了一层碳纳米管亲水性聚合物,通过结合碳纳米管水通道作用和亲水性聚合物基团的亲水性能有效改善pvdf膜的疏水性,进一步提高pvdf膜在实际生产中的应用,且通过该方法得到的pvdf膜表面接枝率、接枝密度、水通量大幅提升。
为了实现上述目的,本发明所采用的技术方案是:一种pvdf膜表面点击碳纳米管分离膜的制备方法,包括以下几个步骤:
(1)摩尔比为0.05-0.5:0.002-0.02:0.5-5的十二硫醇、相转移催化剂、丙酮在氮气气氛和冰浴条件下经磁力搅拌混合均匀,然后滴加饱和naoh溶液1,其中naoh与十二硫醇的摩尔比为0.05-0.5:0.05-0.5,继续搅拌至均匀;再滴加丙酮和cs2混合溶液,其中丙酮、cs2与十二硫醇的摩尔比为0.083-0.83:0.5-5:0.05-0.5,滴加结束后继续搅拌20-30min,然后继续冰浴下滴加三氯甲烷和饱和naoh溶液2,其中三氯甲烷、饱和naoh溶液2中的naoh与十二硫醇的摩尔比为0.075-0.75:0.25-2.5:0.05-0.5),滴加结束后于室温下搅拌反应8-16h,反应结束后加入体积比为35-350:15-150的超纯水和浓hcl充分搅拌后抽滤,然后用有机溶剂提纯和重结晶,再经真空干燥18-36h后得到产物1。
(2)将质量比为0.5-5:0.5-5:0.25-2.5的步骤1所得到的产物1、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(edc-hcl)、4-二甲氨基吡啶(dmap)加入到无水二氯甲烷中并控制体系的质量浓度为0.5-5g/l,然后超声分散30-60min,再在冰浴条件下加入炔丙醇,炔丙醇与产物1的质量比为0.28-2.85:0.5-5,然后室温下反应36-72h,反应结束后依次用hcl、nahco3、nacl溶液洗涤提纯得到产物2,洗涤提纯目的主要是除去未反应的反应物和副产物,其中hcl、nahco3溶液的浓度分别优选3mol/l、1mol/l,nacl溶液优选饱和nacl溶液。
(3)将摩尔比为1-10:20-200:0.002-0.02的步骤2所得的产物2、聚合物单体、偶氮二异丁腈(aibn),在有机溶剂中充分溶解并混合均匀,有机溶剂在体系中的摩尔浓度为0.15mol/l,然后使反应体系在真空氮气氛围下反应,反应温度为30-80℃,时间3-24h,反应结束后得到亲水性聚合物。
(4)向无机酸中加入碳纳米管,(优选的,碳纳米管的加入量为0.017g/ml无机酸),然后在50-80℃(优选60℃)下超声处理0-4h,60-120℃(优选80℃)下加热回流0-8h,反应结束后,用超纯水经高速离心洗涤多次至溶液变为中性,然后取出固体物并于60-120℃真空干燥24h得到产物3,进而完成对碳纳米管的酸化处理,并将碳纳米管长度调整至20微米以下。
(5)将步骤(4)制得的产物3和巯基化硅烷偶联剂分散在无水乙醇中,优选的,(产物3加入量为每ml无水乙醇中加入0.003-0.01g产物3,巯基化硅烷偶联剂与无水乙醇的体积比为0.1-2:10-100),然后在60-120℃(优选70℃)加热回流反应6-12h,反应结束后抽滤洗涤得到产物4,进而实现在碳纳米管上接枝巯基基团。
(6)将质量比为0.1-1:3-30:10-100的步骤5得到的产物4、pvdf、有机溶剂混合并进行超声处理,超声处理30-100min后于40-100℃(优选60℃)下磁力搅拌反应12-24h,然后真空脱泡(脱泡时间优选2h)后用刮膜器均匀刮制50-300μm厚的膜,然后将膜置于超纯水中进行非溶剂相转化后得到pvdf/碳纳米管基膜。
(7)将质量比为1-3:0.3-2的步骤6所得到的pvdf/碳纳米管基膜和步骤(3)制得的亲水性聚合物分散在无水四氢呋喃中,并控制在无水四氢呋喃中pvdf/碳纳米管基膜的质量浓度为1g/ml-10g/ml然后再加入光引发剂,并控制pvdf/碳纳米管基膜与光引发剂的质量比为1-3:0.05-0.3,然后在紫外光照射下反应3-6h,反应结束后将产物于40-80℃(优选60℃)真空干燥(优选24h)得到pvdf膜表面点击碳纳米管分离膜(也可称为pvdf点击碳纳米管分离膜)。
步骤3中,所述聚合物单体为甲基丙烯酸甲酯(mma)、n-异丙基丙烯酰胺、丙烯酸、聚乙二醇、n,n-二甲基丙烯酰胺、二甲基硅氧烷、甲基丙烯酸丁酯中的一种。
进一步的,步骤(1)中,所述相转移催化剂为四正丁基溴化铵、氯化三辛基甲基铵、苄基三乙基氯化铵中的一种。
进一步的,步骤(1)中,所述有机溶剂为异丙醇、甲醇、正丁醇、正己烷中的一种或几种。
进一步的,步骤(3)中,所述有机溶剂为苯甲醚、二甲基甲酰胺、二甲基亚砜、甲苯、1.4-二氧六环、四氢呋喃中的一种。
更好控制碳纳米管的长度和酸化程度,进一步的,步骤4中,所述无机酸为浓hno3、浓h2so4或两者混合而成。
为了接枝效率密度较高,进一步的,步骤5中,所述巯基化硅烷偶联剂为γ-巯丙基甲基二甲氧基硅烷、γ-巯丙基三甲氧基硅烷、γ-巯丙基甲基二乙氧基硅烷中的一种。步骤6中,所述pvdf牌号为761、741、6010、6020、921、5000中的一种,所述有机溶剂为dmac、dmf、nmp、dmso中的一种。步骤7中,所述光引发剂为1173、184、tpo、907中的一种。
基于上述技术方案,本发明所取得的有益的技术效果为:1、通过碳纳米管和pvdf共混制备了含有巯基的pvdf基膜,通过点击化学将pvdf基膜上的巯基和亲水性聚合物上的炔基相互反应,这样接枝效率换和接枝密度更高。2、选用短长度(20微米以下)碳纳米管更有利于碳纳米管的水通道作用在pvdf膜中体现,结合碳纳米管的水通道作用和亲水性聚合物基团的亲水性能,拓宽了pvdf膜在水处理膜领域的应用。
附图说明
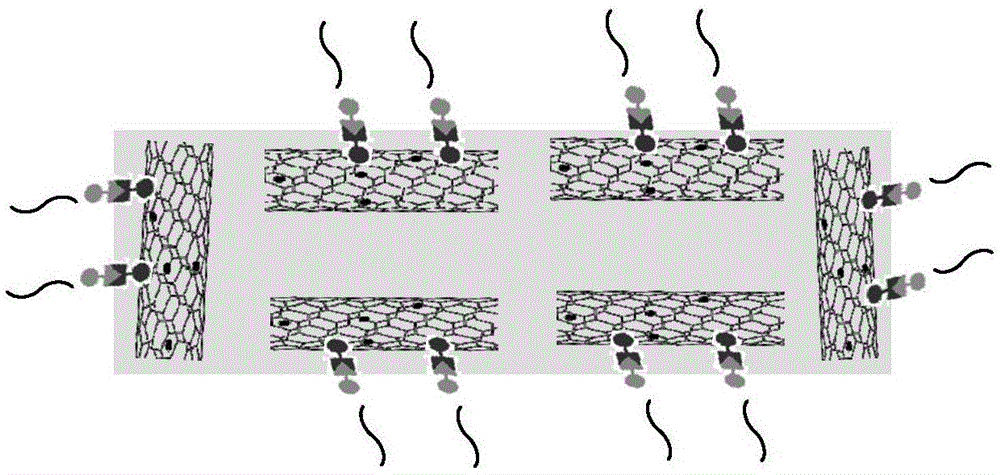
图1为制备得到的pvdf膜表面点击碳纳米管分离膜结构示意图;
图2为实施例1中改性前后pvdf膜接触角变化示意图;
图3为实施例2中改性前后pvdf膜接触角变化示意图;
图4为实施例3中改性前后pvdf膜接触角变化示意图。
具体实施方式
本发明下面结合实施例作进一步详述:以下实施例中所用有机试剂规格均为分析纯ar,dmac、炔丙醇、aibn为国药集团化学有限公司,其余为阿拉丁试剂有限公司。pvdf为阿科玛(arkema)公司、上海三爱富公司、美国3m公司、美国苏威公司中任何一家生产。
实施例1:
本发明专利所述的一种pvdf膜表面点击碳纳米管分离膜的制备方法,包括如下步骤:
第一步:将4.04g十二硫醇、0.26g四正丁基溴化铵、10ml丙酮加入到三口烧瓶中,然后在氮气气氛和冰浴条件下,边进行磁力搅拌边向其中滴加1.68g饱和naoh溶液并继续搅拌至均匀,然后用针筒向其中滴加1.525g二硫化碳和2.015g丙酮的均匀混合溶液,滴加结束后继续搅拌20min后继续冰浴下滴加3.565g氯仿和8.0g饱和naoh溶液,于25℃下反应12h,反应结束后加入50ml超纯水和18ml浓度为12mol/l的浓盐酸充分搅拌后抽滤,然后分别用异丙醇和正己烷提纯重结晶(用异丙醇溶解过滤未溶解的固体,用正己烷重结晶溶液,高温溶解低温结晶得到产物),再于室温下真空干燥24h得到产物1。
第二步:2g产物1、1.72g的edc-hcl和0.67g的dmap溶解在20ml无水二氯甲烷中,超声分散40min后,在冰浴下向其中滴加0.636ml炔丙醇,然后室温下反应48h,反应结束后依次用3mol/lhcl溶液、1mol/lnahco3溶液和饱和nacl溶液洗涤提纯,除去未反应的反应物和副产物,得到产物2。
第三步:2.8gmma单体、4.6mgaibn和56.25mg产物2加入到20ml无水苯甲醚中,经充分溶解并混合均匀后,于真空氮气氛围下反应,反应温度为60摄氏度,反应时间为10h,制得亲水性聚合物。
第四步:将1g碳纳米管加入到45ml浓硝酸中,60℃超声处理30min,80℃下回流6h,反应结束后多次加入超纯水并反复高速离心洗涤至溶液变为中性,然后取出固体物并于60℃真空干燥24h得到产物3。
第五步:将0.15g产物3和0.3mlγ-巯丙基甲基二乙氧基硅烷分散在50ml无水乙醇中,然后在70℃加热回流反应12h,反应结束后抽滤洗涤得到产物4。
第六步:将0.25g产物4、10gpvdf(牌号761)、35gdmac,超声处理40min后于60℃下磁力搅拌反应24h,然后真空脱泡2h后用刮膜器均匀刮膜(膜厚为100μm)后置于超纯水中进行非溶剂相转化后得到pvdf/碳纳米管基膜。
第七步:将亲水性聚合物0.5g、1.5gpvdf/碳纳米管基膜,分散在40ml无水四氢呋喃中,加入0.1g光引发剂184,紫外光照射下反应6h,反应结束后将产物于60℃真空干燥24h,得到pvdf膜表面点击碳纳米管分离膜。。
实施例2
本发明专利所述的一种pvdf膜表面点击碳纳米管分离膜的主要制备工艺方法,包括如下步骤:
第一步:将10.095g十二硫醇、0.645g氯化三辛基甲基铵、24.05g丙酮加入到三口烧瓶中,然后在氮气气氛和冰浴条件下,边进行磁力搅拌边向其中滴加2.095g饱和naoh溶液并继续搅拌至均匀,然后用针筒向其中滴加3.8025g二硫化碳和5.045g丙酮的均匀混合溶液,滴加结束后继续搅拌20min后继续冰浴下滴加8.91g氯仿和10g饱和naoh溶液,于25摄氏度下反应16h,反应结束后加入75ml超纯水和25ml浓度为12mol/l的浓盐酸充分搅拌后抽滤,然后分别用甲醇和正己烷提纯重结晶,再于室温下真空干燥24h得到产物1。
第二步:3.5g产物1、2.89g的edc-hcl和0.99g的dmap溶解在28ml无水二氯甲烷中超声分散50min,在冰浴下向其中滴加0.965ml炔丙醇,然后室温下反应54h,反应结束后依次用3mol/lhcl溶液、1mol/lnahco3溶液和饱和nacl溶液洗涤提纯,除去未反应的反应物和副产物,得到产物2。
第三步:3.3gnipam单体4.92mgaibn和67.05mg产物2加入到15ml的1.4-二氧六环中,经充分溶解并混合均匀后,于真空氮气氛围下反应,反应温度为60摄氏度,反应时间为24h,制得到亲水性聚合物。
第四步:将1g碳纳米管加入到15ml浓硝酸和45ml浓硫酸的混合溶液中,60℃超声处理4h,80℃下回流1h,反应结束后多次加入超纯水并反复高速离心洗涤至溶液变为中性,然后取出固体物并于80℃真空干燥24h得到产物3。
第五步:0.5g产物3和1.2mlγ-巯丙基三甲氧基硅烷分散在70ml无水乙醇中,然后在70℃加热回流反应12h,反应结束后抽滤洗涤得到产物4。
第六步:0.45g产物4、15gpvdf(牌号741)、55gdmac,超声处理40min后60℃下磁力搅拌24h,真空脱泡2h后用刮膜器均匀刮膜(膜厚度为150μm)后置于超纯水中进行非溶剂相转化后得到pvdf/碳纳米管基膜。
第七步:将亲水性聚合物1.5g、2.5gpvdf/碳纳米管基膜,分散在60ml无水四氢呋喃中,加入0.23g光引发剂1173,紫外光照射下反应4h,反应结束后将产物于60℃真空干燥24h,得到pvdf膜表面点击碳纳米管分离膜。
实施例3
本发明专利所述的一种pvdf膜表面点击碳纳米管分离膜的主要制备工艺方法,包括如下步骤:
第一步:将20.30g十二硫醇、1.71g氯化三辛基甲基铵、55.6g丙酮加入到三口烧瓶中,然后在氮气气氛和冰浴条件下,边进行磁力搅拌边向其中滴加8.86g饱和naoh溶液继续搅拌至均匀,然后用针筒向其中滴加7.8g二硫化碳和10.2g丙酮的均匀混合溶液,滴加结束后继续搅拌20min后继续冰浴下滴加17.5g氯仿和36.9g饱和naoh溶液,于25摄氏度下反应16h,反应结束后加入150ml超纯水和50ml浓度为12mol/l的浓盐酸充分搅拌抽滤,然后分别用甲醇和正己烷提纯重结晶,再于室温下真空干燥24h得到产物1。
第二步:4g产物1、2.56g的edc-hcl和0.88g的dmap溶解在25ml无水二氯甲烷中超声分散60min,在冰浴下向其中滴加0.866ml炔丙醇,然后室温下反应72h,反应结束后依次用3mol/lhcl溶液、1mol/lnahco3溶液和饱和nacl溶液洗涤提纯,除去未反应的反应物和副产物,除去未反应的反应物和副产物,得到产物2。
第三步:5gnipam单体7.257mgaibn和72.57mg产物2加入到25ml四氢呋喃中,经充分溶解并混合均匀后,于真空氮气氛围下反应,反应温度为60摄氏度,反应时间为24h,制得到亲水性聚合物。
第四步:将1g碳纳米管加入到30ml浓硝酸和30ml浓硫酸的混合溶液中,60℃超声处理2h,80℃下回流4h,反应结束后多次加入超纯水并反复高速离心洗涤至溶液变为中性,然后取出固体物并于100℃真空干燥24h得到产物3。
第五步:1g产物3和2mlγ-巯丙基三甲氧基硅烷分散在100ml无水乙醇中,然后在70℃加热回流反应12h,反应结束后抽滤洗涤得到产物4。
第六步:0.5g产物4、20gpvdf(牌号6010)、75gdmac,超声处理60min后60℃下磁力搅拌24h,真空脱泡2h后用刮膜器均匀刮膜(膜厚度为150μm)后置于超纯水中进行非溶剂相转化后得到pvdf/碳纳米管基膜。
第七步:将亲水性聚合物2g、3gpvdf/碳纳米管基膜,分散在80ml无水四氢呋喃中,加入0.3g光引发剂907,紫外光照射下反应6h,反应结束后将产物于60℃真空干燥24h,得到pvdf膜表面点击碳纳米管分离膜。
效果实施例
试验一
通过接触角测量仪在室温下测量点击前后pvdf膜的接触角,拍照后利用三点法标定膜的接触角,通过测量得到通过表面点击后pvdf膜的接触角降低至55.6℃。