一种MoV基多元金属氧化物的快速制备方法与流程
本发明涉及金属氧化物制备方法,特别是涉及一种mov基多元金属氧化物的快速制备方法。
背景技术:
mov基多元金属氧化物是以钼、钒两种元素为基体元素,与碲、铌、锑、钽、铬、铁、铈、钨、镓、铋、钾、钴等其他金属或非金属元素化合形成的多元复合金属氧化物(cmo),是一类极为重要的非均相催化剂,在催化烃类选择性氧化脱氢反应中具有不可替代的作用。如,乙烷脱氢制备乙烯,丙烷或丙烯选择性氧化制备丙烯醛或丙烯酸,丁烷或丁烯选择性氧化制备甲基丙烯醛等。在这些反应中,mov基多元金属氧化物的晶相组成(m1相、m2相和(v0.07mo0.93)5o14等)及含量对其催化性能具有显著的影响。而这些晶相结构在很大程度上又取决于所采用的制备方法。
到目前为止,制备mov基多元金属氧化物广泛采用的方法包括共沉淀法(又称浆液法)和水热法两种。其中,共沉淀法是使用较广泛的制备mov基多元金属氧化物的方法(indengchemres2014,53:1775-1786;jmolcatala:chem2014,392:61-66;appliedcatalysisa:general2018,549:152-160;topicsincatalysis2017,60:1401-1407),但其操作步骤极为复杂,涉及了4~5种不同前驱体溶液的配制、4~5个不同温度的控制,整个制备过程大约需要20~30h。在上述操作中每一步都会对催化剂的晶相结构以及催化性能产生重大的影响,使得该方法产生的晶相组成难以控制,产物催化性能的重复性较差。水热法是在共沉淀法之后用于制备mov基多元金属氧化物(appliedcatalysisa:general2014,474:3-9;appliedcatalysisa:general2016,515:179-189;angewandtechemie2016,128:9019-9023),相对于共沉淀法而言,尽管水热法在产物晶相组成控制方面有了一定的改善,但其需要更长的制备时间,整个过程大约需要68~102h。除共沉淀和水热法外,也有文献采用回流法制备了movtenbo催化剂(jmolcatala:chem2011,342-343:50-57),然而仅回流反应时间就需要1~14天。
由上可知,目前的制备方法都存在操作复杂、制备时间过长等问题,严重制约了该类催化剂的应用及发展。综上所述,亟需开发一种简单易行、快速的mov基多元金属氧化物制备方法。
技术实现要素:
本发明的目的在于提供一种mov基多元金属氧化物的快速制备方法,该方法采用超(近)临界流体技术制备mov基多元金属氧化物,操作简单易行、反应时间短,且可通过改变反应条件实现产物晶相组成的调控。
本发明的目的是通过以下技术方案实现的:
一种mov基多元金属氧化物的快速制备方法,具体制备步骤为,
首先将mo源、v源以及其他金属元素前驱物按照一定的摩尔比溶解或分散于去离子水中,混合均匀后加入到反应器中并搅拌,将反应器密闭后,将体系抽真空0~5min,然后将反应器温度加热至250~500℃,使其反应压力保持在0.5~25mpa,在该状态下反应0.1~2.0h,然后将反应器进行冷却,混合液经过分离、洗涤、干燥、煅烧后得到通式为moavbxcydzeo的多元金属氧化物;
mo源为钼酸铵;
v源为钒酸铵、硫酸氧钒中的一种及以上;
其他金属元素前驱物为相应金属的盐、氧化物或含氧酸中的一种及以上。
所述的一种mov基多元金属氧化物的快速制备方法,通式moavbxcydzeo中的x为te,y为nb,z指的是w、ga、bi、k、cr、fe、ce、co、ni、mn、b、p元素中的一种或两种。
所述的一种mov基多元金属氧化物的快速制备方法,通式moavbxcydzeo中的a、b、c、d和e分别为各金属元素的摩尔比值,具体为1、0.1~0.5、0~0.3、0~0.2、0~0.2。
本发明的优点与效果是:
本发明采用超(近)临界流体的方法进行mov基多元金属氧化物的快速制备,反应时间为0.1~2.0h,与传统的共沉淀法和水热法相比,本技术方法不仅大大地缩短了制备时间、简化了制备步骤,而且还能通过调整反应条件对制备产物的晶相组成和含量进行有效的调控。
附图说明
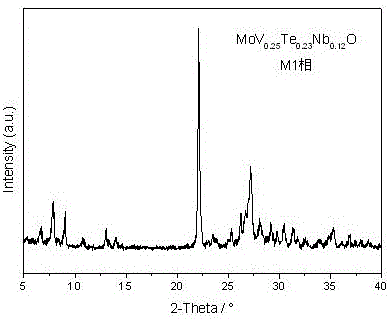
图1为实施例1制得的mov0.25te0.23nb0.12o(m1相)的xrd谱图;
图2为实施例2制得的v0.95mo0.97o5的xrd谱图;
图3为实施例3制得的mov0.25te0.23nb0.12o产物(m1相+m2相)的xrd谱图;
图4为实施例5制得的mov0.3te0.23nb0.12k0.05o(m1相)的xrd谱图;
图5为实施例5制得的v0.12mo0.88o2.94的xrd谱图;
图6为实施例6制得的mov0.25te0.23nb0.12o产物(m1相+m2相+(v0.07mo0.93)5o14)的xrd谱图。
具体实施方式
以下通过实例对本发明做进一步阐述,但不限制本发明。该制备方法的操作步骤为:
首先将mo、v以及其他金属元素相应的盐或含氧酸按照一定的摩尔比溶解或分散于一定体积的去离子水中。混合均匀后加入到反应器中并搅拌,将反应器密闭后,抽真空0~5min,然后将反应器温度加热至250~500℃,使其反应压力保持在0.5~25mpa,并在该状态下反应0.1~2.0h,反应结束后,将反应器冷却降温,取出的反应混合液经过分离、洗涤、干燥、煅烧后得到通式为moavbxcydzeo的多元金属氧化物;
实施例1:
称取四水合钼酸铵(2.23g)、偏钒酸铵(0.36g)、碲酸(0.65g)和水合草酸铌(0.80g)于80℃分散于30ml去离子水中,形成均匀的橘红色浆液,将上述浆液转移至反应器中并搅拌,将反应器密闭并抽真空5min后,加热反应器至400℃,在该温度下反应2h,反应过程中体系压力为4.0mpa。反应结束后,停止加热,自然冷却,收集反应体系,离心分离、洗涤、真空干燥,并于氮气气氛下600℃煅烧2.0h,得到1.88gmov0.25te0.23nb0.12o(m1相)产物。
实施例2:
将1.66g四水合钼酸铵和0.48g偏钒酸铵在80℃下溶于25ml去离子水中,形成橘黄色溶液,将该溶液转移至反应器中并搅拌,反应器密闭后,将其加热至350℃,体系压力为2.5mpa,在该条件下保持1.0h后,停止加热,冷却后取出所得样品,抽滤、洗涤、干燥后得0.57g产物v0.95mo0.97o5。
实施例3:
称取四水合钼酸铵(0.74g)、偏钒酸铵(0.12g)、碲酸(0.22g)和水合草酸铌(0.27g)于80℃分散于30ml去离子水中,形成均匀的橘红色浆液,将上述浆液转移至反应器中并搅拌,将反应器密闭并抽真空5min后,加热反应器至400℃,体系压力为1.4mpa,在该条件下反应1h,反应结束后,停止加热,自然冷却,收集反应体系,抽滤、洗涤、真空干燥,并于氮气气氛下600℃煅烧2.0h,得到0.58gmov0.25te0.23nb0.12o产物(m1相+m2相)。
实施例4:
称取0.71g钼酸铵、0.1413g偏钒酸铵和0.02g硝酸钾加入20ml去离子水中加热溶解成溶液,再将0.211g碲酸和0.251g草酸铌溶于15ml去离子水中,将上述两种溶液混合并搅拌均匀后,转移至反应器中,将反应器密闭后加热至420℃,体系压力为15.6mpa在该条件下保持0.5h后,停止加热,待反应器冷却后,取出反应体系,固体颗粒经离心分离、洗涤、真空干燥后,在氮气气氛下600℃煅烧2.0h,得到mov0.3te0.23nb0.12k0.05o产物(m1相)。
实施例5:
称取四水合钼酸铵2.23g溶解于40ml80℃的去离子水中,在搅拌状态下,再向其中加入0.37g硫酸氧钒,继续搅拌溶解形成橘黄色澄清溶液,将该溶液转移至反应器中并搅拌,反应器密闭后,将其加热至380℃,体系压力为17.3mpa,在该条件下保持1.5h后,停止加热,冷却后取出所得样品,抽滤、洗涤、干燥后得到黑色产物v0.12mo0.88o2.94。
实施例6:
将2.21g四水合钼酸铵、0.36g偏钒酸铵和0.66g碲酸依次加入至25ml去离子水中,于80℃下搅拌溶解,形成酒红色透明溶液,再向上述溶液中加入0.81g水合草酸铌,继续在80℃下搅拌10min,体系形成橘黄色浆液,将上述浆液转移至反应器中并搅拌,将反应器密闭后,加热反应器至400℃,在该温度下反应2.0h,反应过程中体系压力为3.0mpa。反应结束后,停止加热,自然冷却,收集反应体系,抽滤、洗涤、真空干燥,并于氮气气氛下600℃煅烧2.0h,得到蓝黑色产物m1相+m2相+(v0.07mo0.93)5o14。
- 还没有人留言评论。精彩留言会获得点赞!