一种铌酸钾基光催化剂的制备方法及其应用与流程
本发明涉及能源技术领域,尤其是涉及一种铌酸钾基光催化剂的制备方法及其应用。
背景技术:
将太阳能转化为氢能是解决能源短缺和环境污染问题的一条潜在途径。在各种技术中,光催化制氢是实现这一转变的最有希望的方法之一,其中新颖和高度活性的光催化剂的制备是至关重要的。一种理想的光催化剂应该是高效、稳定和廉价的。
由于铌酸钾光催化剂结构稳定、形态可调、光催化活性较好,因此受到越来越多研究者的关注。通过改变表面积和亲水性,发现铌酸钾的结构和形貌能够得到调制从而对其光催化活性有重要影响。因此,可以通过调节铌酸钾光催化剂的结构和形态来调整它们的光催化活性。专利cn201010155915提出了一种制备铌酸钾纳米管催化剂的方法,并可通过在铌酸钾纳米管上负载金属如铂等提高其光催化性能。但是该方法所制备的铌酸钾催化剂中,金属只是被负载在催化剂上,没有掺杂在催化剂中直接影响催化剂的能带结构,同时所采用的金属铂为贵金属,价格较贵。掺杂是降低k4nb6o17带隙的最常用的方法,因为它可以引起半导体本征的电子和能带结构变化。然而,将金属离子掺杂到k4nb6o17的晶格中可能会导致热或晶体不稳定,增加电荷载体复合中心,从而降低光催化活性。
技术实现要素:
本发明的目的就是为了克服上述现有技术存在的缺陷而提供一种铌酸钾基光催化剂及其应用,显著增强铌酸钾基光催化剂的制氢活性。
本发明的目的可以通过以下技术方案来实现:
一种铌酸钾基光催化剂的制备方法,包括以下步骤:
(1)将nb2o5分散于koh溶液中得到nb2o5分散液,经过高压高温反应得到反应液,将该反应液过滤得到上层清液;
(2)向所述上层清液中加入尿素溶液,经过高压高温反应得到反应液,将该反应液过滤得到滤饼,将滤饼进行洗涤、干燥得到所述铌酸钾基光催化剂。
本发明提供了一种自掺杂的铌酸钾基光催化剂,研究发现,自掺杂是一种有效且有前景的方法来拓展半导体光催化剂的吸收。
本发明的制备方法得到的铌酸钾光催化剂可以单独使用,由于具有多孔的结构和较高的比表面积,因此,本发明的催化剂在单独使用时,其催化性能优于现有技术中合成方法得到的铌酸钾。
并且,发明制备铌酸钾的方法过程简单,仅需将反应原料分散、混合,经过两次高温高压反应就可以得到产品,反应时间较短,整个过程反应时间仅为38小时,而对比文件cn201010155915中,酸化和剥落两步时间共计为7天,说明本发明大大缩短了合成催化剂的时间,大大提高了生产效率。
所述步骤(1)中,所述koh溶液的浓度为1~5mol·l-1,优选为2~4mol·l-1。
所述步骤(1)中,所述nb2o5分散液中nb2o5的浓度为0.005~0.1g·ml-1,优选为0.01~0.05g·ml-1。
所述步骤(1)中,高温高压反应的反应温度为160~200℃,优选为170~190℃;160.7~175.6kpa,优选为164.5~171.9kpa;反应时间为8~12小时,优选为9~11小时。
步骤(2)中,所述尿素溶液的浓度为0.05~0.2g·ml-1,优选为0.1~0.15g·ml-1;所述尿素溶液的体积与步骤(1)中koh溶液的体积比为1:1~1:4,优选体积比为1:2~1:3。
步骤(2)中,高温高压反应的反应温度为200~240℃,优选为210~230℃;反应压力为175.6~190.5kpa,优选为179.3~186.7kpa;反应时间为18~30小时,优选为20~26小时。步骤(2)中的洗涤方法为采用去离子水和无水乙醇洗涤数次;干燥温度为60℃。
原料的配比,浓度,反应时间,反应温度和反应压力;不采用本发提供的工艺参数,将会影响样品的微观尺度和形貌,降低其催化性能。
本发明进一步提供了一种铌酸钾基光催化剂的制备方法的应用,将铜掺杂到铌酸钾基光催化剂中制备得到铜掺杂的铌酸钾基光催化剂。通过非贵金属铜纳米粒子掺杂铌酸钾以提高其光催化制氢活性。
该制备方法包括以下步骤:
(1)将所述铌酸钾基光催化剂分散于溶剂水中得到铌酸钾基光催化剂分散液;
(2)向所述铌酸钾基光催化剂分散液中加入可溶性铜盐溶液,室温搅拌得到混合溶液;
(3)氮气气氛下向所述混合溶液中加入n2h4·h2o,进行还原反应,将反应后的溶液进行过滤、洗涤、干燥得到所述铜掺杂的铌酸钾基光催化剂。
本发明进一步在铌酸钾基光催化剂上掺杂了cu,cu在铌酸钾催化剂上形成了铜纳米颗粒,cu作为助剂进一步提高了铌酸钾主催化剂的制氢效果。
在紫外/紫外-可见光照射下激发k4nb6o17微米花产生了电子/空穴对,然后将光生电子转移到导带(cb)上,在此过程中,一些电子被nb5+捕获,在cunps存在下迅速转化为nb4+,使得电子/空穴对的分离更加有效。因此,复合微米花中的cunps至关重要,它具有双重作用:(i)是nb5+向nb4+转化的催化剂,完成k4nb6o17的自掺杂;(ii)作为光催化制氢的助催化剂和活性中心。在此之后,光生电子转移到活性中心,h2o被还原产生h2。牺牲剂(甲醇)消耗了k4nb6o17微米花vb上积累的空穴。因此,得益于cunps对k4nb6o17微观结构的调控以及cunps与k4nb6o17微米花之间强的界面接触,使得光生电子倾向于从k4nb6o17光催化剂迁移到cunps为吸附溶液中的质子提供了活性位点微米花;即cunps与k4nb6o17微米花之间有效的电子转移,显著延长了光激发载流子的寿命,从而提高了光催化制氢活性。
通过表征,我们发现cu以零价态纳米颗粒形式存在,通过在第(3)步中,在氮气氛围下还原,能够很好地使cu2+还原成零价态的cu纳米颗粒。cu纳米颗粒的双重作用使得催化剂的催化性能,避免加入贵金属铂,大大降低了催化剂的成本。
所述铜掺杂的铌酸钾基光催化剂中cu的质量分数为0.2%~3%,优选为0.4%;
所述可溶性铜盐溶液选自醋酸铜溶液,cucl2溶液,cu(no3)2溶液或cuso4溶液中的一种或几种;
只要是在水溶液中可以很好的溶解电离出铜离子的铜盐均可使用。
步骤(1)中,所述铌酸钾基光催化剂分散液的浓度为0.2~2mg·ml-1,优选1~1.5mg·ml-1;
步骤(2)中可溶性铜盐溶液的浓度为0.005~0.075mol·l-1;
步骤(3)中,所述n2h4·h2o与所述可溶性铜盐的质量比为0.24~0.5。
所述还原反应的温度为40~98℃,优选为90℃,反应时间为2~8小时,优选为5小时。
步骤(3)中的洗涤方法为采用去离子水和无水乙醇洗涤数次;干燥温度为60℃。
原料的配比,浓度,反应时间,反应温度和反应压力;不采用本发提供的工艺参数,将会影响样品的微观尺度和形貌,得不到合适分散效果的铜纳米颗粒,降低其催化性能。
与现有技术相比,本发明具有以下有益效果:
(1)本发明的制备方法得到了一种显著增强制氢活性的新型、高效的铌酸钾基光催化剂;
(2)本发明将非贵金属cu掺杂进铌酸钾基光催化剂中,进一步提高了其光催化性能,并且相比于贵金属掺杂,本发明的原料价格低廉、来源广泛,大大降低了生产成本;
(3)本发明的制备工艺路径简单、容易操作、反应原料易得,容易扩大生产。
(4)避免加入贵金属如铂等助催化剂,大大降低原料成本。
(5)小尺寸、单分散铜粒子在铌酸钾微米花瓣上的负载,既避免cu的脱落,又提高催化剂的稳定性和催化活性,而且大颗粒的催化剂利于回收。
附图说明
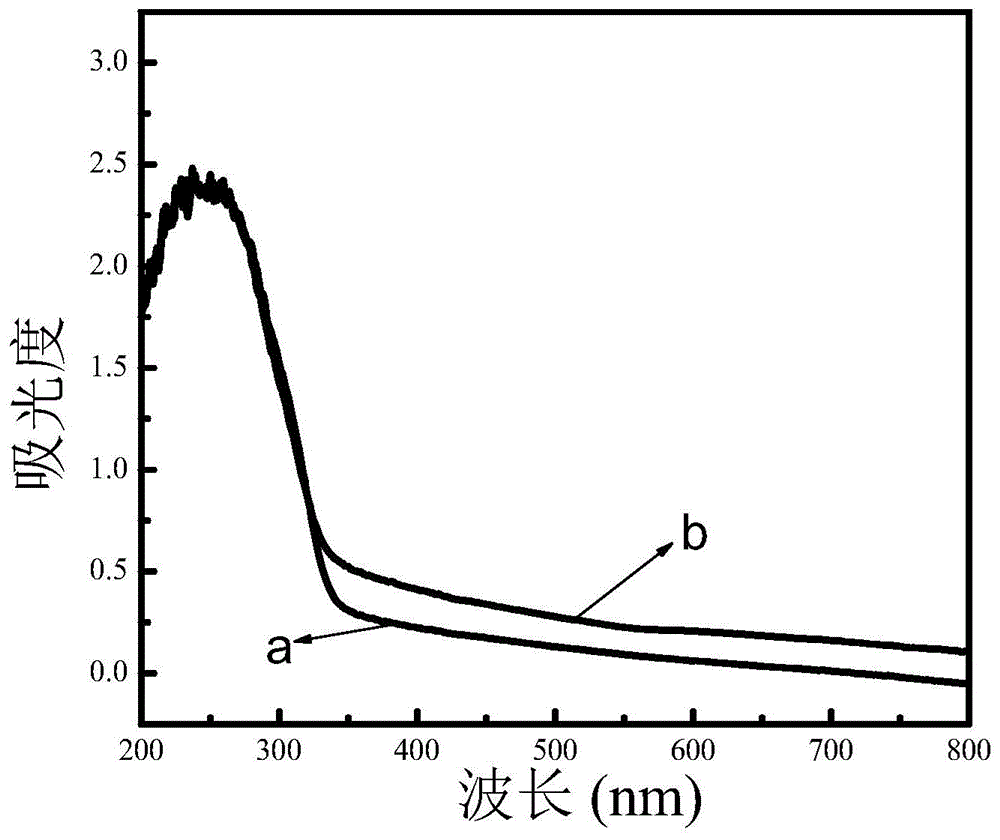
图1为0.4%cu/k4nb6o17微米花经紫外-可见光照射前(a)和照射后(b)的紫外-可见漫反射光谱;
图2为0.4%cu/k4nb6o17微米花经紫外-可见光照射前(a)和照射后(b)的nb3d高分辨率xps谱;
图3为cu/k4nb6o17复合微米花的透射电镜照片;
图4为图3中铜元素分布照片。
具体实施方式
下面结合具体实施例对本发明进行详细说明。以下实施例将有助于本领域的技术人员进一步理解本发明,但不以任何形式限制本发明。应当指出的是,对本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进。这些都属于本发明的保护范围。
实施例1
本实施例为一种铌酸钾基光催化剂,制备方法为:
将0.3gnb2o5分散在30ml的3moll-1koh溶液中,然后在180℃下的不锈钢高压釜中反应约10小时,反应压力为171.9kpa。从高压釜中取出9ml澄清溶液,并将2g尿素和18ml去离子水加入其中。然后,将混合物装入不锈钢高压釜中,并在220℃加热24小时。过滤收集目标产物,分别用去离子水和无水乙醇洗涤数次,并在60℃下干燥,得到k4nb6o17。
实施例2
本实施例为一种铜掺杂的铌酸钾基光催化剂,铜的负载量为0.38%wt,制备方法为:
取实施例1中的铌酸钾基光催化剂,将100mgk4nb6o17分散到100ml去离子水中,向分散体中加入0.6ml0.01·moll-1cucl2溶液,然后在室温下搅拌24小时。在氮气气氛下,向上述混合物中缓慢加入0.2μln2h4·h2o,并在90℃继续反应5小时。然后,过滤得到目标产物cu/k4nb6o17,分别用去离子水和乙醇冲洗数次。所得固体在60℃的真空烘箱中干燥12小时,得到铜掺杂的铌酸钾基光催化剂。
实施例3
本实施例为一种铌酸钾基光催化剂,制备方法为:
将0.3gnb2o5分散在30ml的3mol·l-1koh溶液中,然后在160℃下的不锈钢高压釜中反应约8小时,反应压力为171.9kpa。从高压釜中取出9ml澄清溶液,并将2g尿素和18ml去离子水加入其中。然后,将混合物装入不锈钢高压釜中,并在220℃加热30小时。过滤收集目标产物,分别用去离子水和无水乙醇洗涤数次,并在60℃下干燥,得到k4nb6o17。
实施例4
本实施例为一种铜掺杂的铌酸钾基光催化剂,铜的负载量为0.38%wt,制备方法为:
取实施例3中的铌酸钾基光催化剂,将100mgk4nb6o17分散到100ml去离子水中,向分散液中加入0.6ml的0.01mol·l-1cucl2溶液,然后在室温下搅拌24小时。在氮气气氛下,向上述混合物中缓慢加入0.2μln2h4·h2o,并在90℃继续反应5小时。然后,过滤得到目标产物cu/k4nb6o17),分别用去离子水和乙醇冲洗数次。所得固体在60℃的真空烘箱中干燥12小时。
实施例5
本实施例为一种铌酸钾基光催化剂,制备方法为:
将0.3gnb2o5分散在30ml的5mol·l-1koh溶液中,然后在180℃下的不锈钢高压釜中反应约12h,反应压力为171.9kpa。从高压釜中取出9ml澄清溶液,并将2g尿素和18ml去离子水加入其中。然后,将混合物装入不锈钢高压釜中,并在220℃加热18小时。过滤收集目标产物,分别用去离子水和无水乙醇洗涤数次,并在60℃下干燥,得到k4nb6o17。
实施例6
本实施例为一种铜掺杂的铌酸钾基光催化剂,铜的负载量为1.54%wt,制备方法为:
取实施例5中的铌酸钾基光催化剂,将100mgk4nb6o17分散到100ml去离子水中,向分散体中加入0.6ml的0.04mol·l-1cucl2溶液,然后在室温下搅拌24小时。在氮气气氛下,向上述混合物中缓慢加入0.8μln2h4·2h2o,并在90℃继续反应5小时。然后,过滤得到目标产物cu/k4nb6o17),分别用去离子水和乙醇冲洗数次。所得固体在60℃的真空烘箱中干燥12小时。
实施例7
本实施例为一种铌酸钾基光催化剂,制备方法为:
将nb2o5分散在30ml的2mol·l-1koh溶液中,使nb2o5在koh溶液中的浓度为0.005g·ml-1,然后在170℃下的不锈钢高压釜中反应约9小时,反应压力为175.6kpa。从高压釜中取出9ml澄清溶液,并将30ml的0.1g·ml-1的尿素水溶液加入其中。然后,将混合物装入不锈钢高压釜中,并在210℃加热26小时,反应压力为186.7kpa。过滤收集目标产物,分别用去离子水和无水乙醇洗涤数次,并在60℃下干燥,得到k4nb6o17。
实施例8
本实施例为一种铜掺杂的铌酸钾基光催化剂,铜的负载量为0.2%wt,制备方法为:
取实施例7中的铌酸钾基光催化剂,将20mgk4nb6o17分散到100ml去离子水中,向分散体中加入0.125ml0.005·moll-1醋酸铜溶液,然后在室温下搅拌24小时。在氮气气氛下,向上述混合物中缓慢加入0.03μln2h4·h2o,并在98℃继续反应2小时。然后,过滤得到目标产物cu/k4nb6o17,分别用去离子水和乙醇冲洗数次。所得固体在60℃的真空烘箱中干燥12小时,得到铜掺杂的铌酸钾基光催化剂。
实施例9
本实施例为一种铌酸钾基光催化剂,制备方法为:
将nb2o5分散在30ml的4mol·l-1koh溶液中,使nb2o5在koh溶液中的浓度为0.05g·ml-1,然后在190℃下的不锈钢高压釜中反应约9小时,反应压力为171.9kpa。从高压釜中取出9ml澄清溶液,并将15ml的0.15g·ml-1的尿素水溶液加入其中。然后,将混合物装入不锈钢高压釜中,并在230℃加热20小时,反应压力为190.5kpa。过滤收集目标产物,分别用去离子水和无水乙醇洗涤数次,并在60℃下干燥,得到k4nb6o17。
实施例10
本实施例为一种铜掺杂的铌酸钾基光催化剂,铜的负载量为3%wt,制备方法为:
取实施例7中的铌酸钾基光催化剂,将150mgk4nb6o17分散到100ml去离子水中,向分散体中加入0.94ml0.075·moll-1cu(no3)2溶液,然后在室温下搅拌24小时。在氮气气氛下,向上述混合物中缓慢加入2.35μln2h4·h2o,并在40℃继续反应8小时。然后,过滤得到目标产物cu/k4nb6o17,分别用去离子水和乙醇冲洗数次。所得固体在60℃的真空烘箱中干燥12小时,得到铜掺杂的铌酸钾基光催化剂。
实施例11
本实施例为一种铌酸钾基光催化剂,制备方法为:
将nb2o5分散在30ml的1mol·l-1koh溶液中,使nb2o5在koh溶液中的浓度为0.1g·ml-1,然后在160℃下的不锈钢高压釜中反应约11小时,反应压力为164.5kpa。从高压釜中取出9ml澄清溶液,并将10ml的0.05g·ml-1的尿素水溶液加入其中。然后,将混合物装入不锈钢高压釜中,并在240℃加热18小时,反应压力为179.3kpa。过滤收集目标产物,分别用去离子水和无水乙醇洗涤数次,并在60℃下干燥,得到k4nb6o17。
实施例12
本实施例为一种铜掺杂的铌酸钾基光催化剂,铜的负载量为0.4%wt,制备方法为:
将200mgk4nb6o17分散到100ml去离子水中,向分散体中加入1.2ml的0.04mol·l-1cuso4溶液,然后在室温下搅拌24小时。在氮气气氛下,向上述混合物中缓慢加入1.6μln2h4·h2o,并在90℃继续反应5小时。然后,过滤得到目标产物cu/k4nb6o17),分别用去离子水和乙醇冲洗数次。所得固体在60℃的真空烘箱中干燥12小时,得到cu/k4nb6o17。
实施例13
本实施例为一种铌酸钾基光催化剂,制备方法为:
将nb2o5分散在30ml的1mol·l-1koh溶液中,使nb2o5在koh溶液中的浓度为0.015g·ml-1,然后在200℃下的不锈钢高压釜中反应约8小时,反应压力为160.7kpa。从高压釜中取出9ml澄清溶液,并将7.5ml的0.2g·ml-1的尿素水溶液加入其中。然后,将混合物装入不锈钢高压釜中,并在200℃加热30小时,反应压力为175.6kpa。过滤收集目标产物,分别用去离子水和无水乙醇洗涤数次,并在60℃下干燥,得到k4nb6o17。
实施例14
为了说明我们制备的目标光催化剂的优势,对比制备了负铜前、后以及同样条件下负铂催化剂,本实施例中的铌酸钾催化剂采用实施例1中的铌酸钾催化剂,并采用该铌酸钾催化剂进一步通过以下方法制备铜掺杂的铌酸钾基光催化剂以及铂掺杂的铌酸钾基光催化剂。
其中,一种铜掺杂的铌酸钾基光催化剂,制备方法为:
将100mgk4nb6o17分散到100ml去离子水中,向分散体中加入0.6ml的0.01mol·l-1cucl2溶液,然后在室温下搅拌24小时。在氮气气氛下,向上述混合物中缓慢加入0.2μln2h4·h2o,并在90℃继续反应5小时。然后,过滤得到目标产物cu/k4nb6o17),分别用去离子水和乙醇冲洗数次。所得固体在60℃的真空烘箱中干燥12小时,得到cu/k4nb6o17。
其中,一种铂掺杂的铌酸钾基光催化剂,制备方法为:
将100mgk4nb6o17分散到100ml去离子水中,向分散体中加入0.6ml的0.01mol·l-1h2ptci6·6h2o溶液,然后在室温下搅拌24小时。在氮气气氛下,向上述混合物中缓慢加入0.2μln2h4·h2o,并在90℃继续反应5小时。然后,过滤得到目标产物cu/k4nb6o17),分别用去离子水和乙醇冲洗数次。所得固体在60℃的真空烘箱中干燥12小时,得到pt/k4nb6o17。
对实施例1中的铌酸钾催化剂以及铜掺杂的铌酸钾基光催化剂、铂掺杂的铌酸钾基光催化剂的光催化制氢效果进行测试。
催化剂的活性评价
测试条件为:通过cel-sp2n水分解系统(中教金源)进行样品的光催化析氢活性研究。光催化析氢实验主要过程如下:将60mg光催化剂加入到含有甲醇水溶液(60ml,水与甲醇的体积比为4:1)的光催化反应器中,磁力搅拌均匀。将反应器与气体循环系统连接,通过在线气相色谱仪(nax沸石柱,高纯度n2作为载气,热导检测器)分析产生的氢气量。根据实验需要,光源是500w汞灯(uv光),300w氙灯带380nm滤光片(可见光),或300w氙灯不带滤光片(uv-vis光)。通过循环水夹套将反应温度保持在约8.5℃。产生的气体自动取样并通过在线气相色谱分析。在光催化反应之前,通过真空泵交替地抽空反应器并用高纯度n2冲洗几次以确保完全除去氧气。
测试结果如表1所示。
表1相同条件下光照6小时制氢量比较
从表1中可以看出,采用本发明的铌酸钾催化剂,制氢量为221.5μmol·g-1,相比于对比文件cn101811044a中的铌酸钾催化剂(制氢量为82.04μmol·g-1),本发明的铌酸钾催化剂的单独催化效率大大提升,是现有技术中铌酸钾催化剂的3倍左右,大大提高了其光催化制氢活性;
另外,对比cu/k4nb6o17和pt/k4nb6o17催化剂,发现在金属助剂负载量相同的条件下,负载价格较为低廉的铜金属的效果反而要远远高于负载贵金属pt,因此,本发明中的铌酸钾尤其适用于负载非贵金属,不仅提高了光催化活性,还大大降低了原料成本。
将本发明中获得催化效果较好的cu/k4nb6o17催化剂与现有技术中的催化剂进行活性比较,如表2所示。
表2本发明与现有技术的催化效果比较
由表2可知,贵金属pt在knb3o8上的沉积是提高光催化活性的有效途径。但贵金属价格昂贵,不利于其实际应用。此外,k4nb6o17与cu2s或cds偶联可大大提高其光催化活性。与这些催化剂相比,cu/k4nb6o17复合微米花显示了较高的催化析氢活性,甚至高于pt/knb3o8和pt/k4nb6o17微米花。重要的是,cu/k4nb6o17中cunps的负载量低于pt/knb3o8中pt的量。虽然cu/k4nb6o17复合微米花的光催化活性略低于cu2s/k4nb6o17,但我们的研究只引入了0.4%的cu。总之,采用廉价金属cunps激发k4nb6o17自掺杂复合微米花的制备和光催化机理,将为设计新型光催化剂、探索助催化剂的作用和光催化机理提供依据。
催化剂的结构表征
对实施例中的k4nb6o17催化剂进行bet吸附表征,计算得到该催化剂的比表面积高达77.7m2/g,较高的比表面积不仅有利于提供更多的催化活性位点,使本实施例中的铌酸钾催化剂具有更高的光催化活性,还有利于cu等金属的分散,从而合成活性更好的掺杂型催化剂。
对cu/k4nb6o17催化剂进行结构表征,获得了0.4%cu/k4nb6o17微米花经紫外-可见光照射前(a)和照射后(b)的紫外-可见漫反射光谱,如图1所示。
当对cu/k4nb6o17微米花的悬浮液照射6小时后,观察到混合物的颜色由白色变为灰黑色,用紫外-可见光光漫反射光谱进行分析,如图1所示,光照后样品在可见光区域的吸光度明显增强,推断有可能发生了自掺杂。进一步通过高分辨率的xps谱分析了光照前后nb价态变化,如图2所示,在207.1ev和209.9ev处观察到典型的nb3d5/2和nb3d3/2峰,经过光照射后,位移至206.9ev和209.7ev,发生了nb5+向nb4+的转变,证明自掺杂催化剂的成功合成。
利用透射电镜对样品的形貌进行分析,从图3可以看出,在k4nb6o17微米花的花片上均匀负载了一些衬度较深的纳米颗粒,尺寸在2-11nm之间,大部分为4-5nm。此外,在图像中没有观察到纳米颗粒的聚集或游离于花外,这说明cunps能被有效负载到k4nb6o17微米花上,且将cunps负载到微米花上可以有效避免cunps的聚集。此外,采用能量过滤透射电镜进一步分析了复合微米花中cunps的分布。如图4所示,其中亮的地方为cu颗粒,可以清晰地观察到铜元素的分布均匀,并且铜颗粒均为纳米级的铜颗粒。这说明cunps已成功掺杂到k4nb6o17微米花中,且cunps的尺寸较小,且分散性好。另外,对实施例中的cu/k4nb6o17催化剂进行bet吸附表征,计算得到该催化剂的比表面积高达111.0m2/g。
以上对本发明的具体实施例进行了描述。需要理解的是,本发明并不局限于上述特定实施方式,本领域技术人员可以在权利要求的范围内做出各种变形或修改,这并不影响本发明的实质内容。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种电极催化材料的制备方法及其在葡萄糖燃料电池中的应用与流程
- 一种负载型Bi2MoO6/Cu(OH)2/石墨烯光催化剂的制备方法与流程
- 一种高效产氢的球状纳米ZnO/ZnCr2O4复合光催化剂的制备方法与流程
- 一种铬酸银/溴化银复合光催化剂的制备方法与流程
- 一种MoS2纳米片复合TiO2纳米片的光催化剂的制备方法与流程
- 一种CdS量子点/Bi2MoO6/石墨烯复合型光催化剂的制备方法与流程
- 一种Cu/ZnO光催化剂的制备方法与流程
- CQDs/TNS和SiQDs/TNS复合光催化剂的制备方法与流程
- 能够有效降解染料废水的Bi2S3/SnS2/Bi2O3三元复合光催化剂的制备方法与制造工艺
- 羟基锡酸钴/石墨烯复合光催化剂的制备方法及其应用与制造工艺