一种用于α,β-不饱和醛/酮选择性加氢的催化剂、制备方法及催化方法与流程
本发明属于选择加氢催化剂技术领域,特别涉及一种用于α,β-不饱和醛/酮选择性加氢的催化剂、制备方法及其催化方法。
背景技术:
α,β-不饱和醛/酮,诸如苄叉丙酮、2-环己烯-1-酮、2-甲基-2-戊烯醛、巴豆醛、3-庚烯-2-酮、2-甲基丙烯醛和肉桂醛等是重要的化工原料及中间体,α,β-不饱和醛的c=o键选择性加氢得到的α,β-不饱和醇可用于精细化学品、香水及制药行业,而α,β-不饱和醛/酮的c=c键选择性加氢得到的饱和醛/酮可制备香料和医药的重要中间体,譬如苯丙醛用于合成抗艾滋病毒化合物。从热力学角度看,c=c键的键能为615kj/mol,c=o键的键能为715kj/mol,因而c=c键比c=o键更容易加氢。但是由于α,β-不饱和醛/酮中的c=c键与c=o键形成共轭体系,使c=o键也很容易被还原,发生c=c键和c=o键的竞争加氢反应,产物往往是α,β-不饱和醛/酮中的c=c键、c=o键以及二者都被加氢饱和的多种物质的混合物。α,β-不饱和醛/酮中的c=c键或c=o键的高效定向地催化加氢一直是学术界和工业界关注的焦点。
技术实现要素:
本发明所解决的技术问题是提供一种高效定向地催化α,β-不饱和醛/酮的c=c键加氢的催化剂,高选择性地进行α,β-不饱和醛/酮加氢,并且能够降低反应温度、压力以及使用绿色溶剂,可降低投资成本、减少能耗,降低对环境的不良影响。
本发明所述用于α,β-不饱和醛/酮选择性加氢的催化剂,所述催化剂用于定向催化所述α,β-不饱和醛/酮中的c=c键的加氢反应;所述催化剂包括活性组分钯以及多孔材料催化剂载体;所述活性组分钯与所述多孔材料催化剂载体的质量比为(0.1~10.0):100;所述多孔材料催化剂载体为尖晶石型结构化合物。
在其中一个实施例中,所述尖晶石型结构化合物为镍钴酸盐或镍钴酸盐复合物。
在其中一个实施例中,所述活性组分钯与所述多孔材料催化剂载体的质量比为(0.5~1.5):100。
本发明还提供一种如上所述的催化剂的制备方法,所述制备方法包括以下步骤:
催化剂浸渍制备:将所述多孔材料催化剂载体与所述活性组分钯的金属盐溶液混合浸渍反应1h~24h,制得催化剂浸渍混合物;
还原处理:将所述催化剂浸渍混合物与还原剂溶液混合反应1h~3h,制得所述催化剂。
在其中一个实施例中,所述活性组分钯的金属盐为四氯钯酸钾、氯化钯、二氯四氨合钯中的任意一种。
在其中一个实施例中,所述活性组分钯的金属盐溶液的摩尔浓度为0.001mol/l~0.10mol/l。
在其中一个实施例中,所述还原剂溶液为硼氢化钠溶液,在所述还原步骤中,活性组分钯的金属盐与硼氢化钠的摩尔比为1:(1~9)。
本发明还提供了一种如上所述的催化剂的催化方法,所述催化方法包括以下步骤:
将α,β-不饱和醛/酮底物、所述催化剂以及溶剂于常压下通入氢气反应,反应温度为30℃~60℃。
在其中一个实施例中,所述催化剂的用量为所述α,β-不饱和醛/酮底物质量的1.0%~1.5%。
在其中一个实施例中,所述α,β-不饱和醛/酮底物包括肉桂醛、苄叉丙酮、2-环己烯-1-酮、2-甲基-2-戊烯醛、巴豆醛、3-庚烯-2-酮、甲位戊基桂醛、α-溴代肉桂醛、异佛尔酮。
上述用于α,β-不饱和醛/酮选择性加氢的催化剂,所述催化剂用于定向催化α,β-不饱和醛/酮中的c=c键的加氢反应;催化剂中的活性组分钯以纳米粒子形式负载在多孔材料催化剂载体上,其粒径细小且均匀,并且高度分布在多孔材料催化剂载体上,从而能够使催化剂的催化活性高、稳定性高,催化c=c键的加氢反应选择性高、转化率高,且催化条件更为温和,能够在常压且较低的温度条件下进行定向加氢反应,并且催化加氢使用的溶剂更加绿色安全,易于回收处理,降低了投资成本、减少能耗,并且降低了对环境的不良影响。
附图说明
为了更清楚地说明本申请实施例或现有技术中的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明中记载的一些实施例,对于本领域普通技术人员来讲,还可以根据这些附图获得其他的附图。
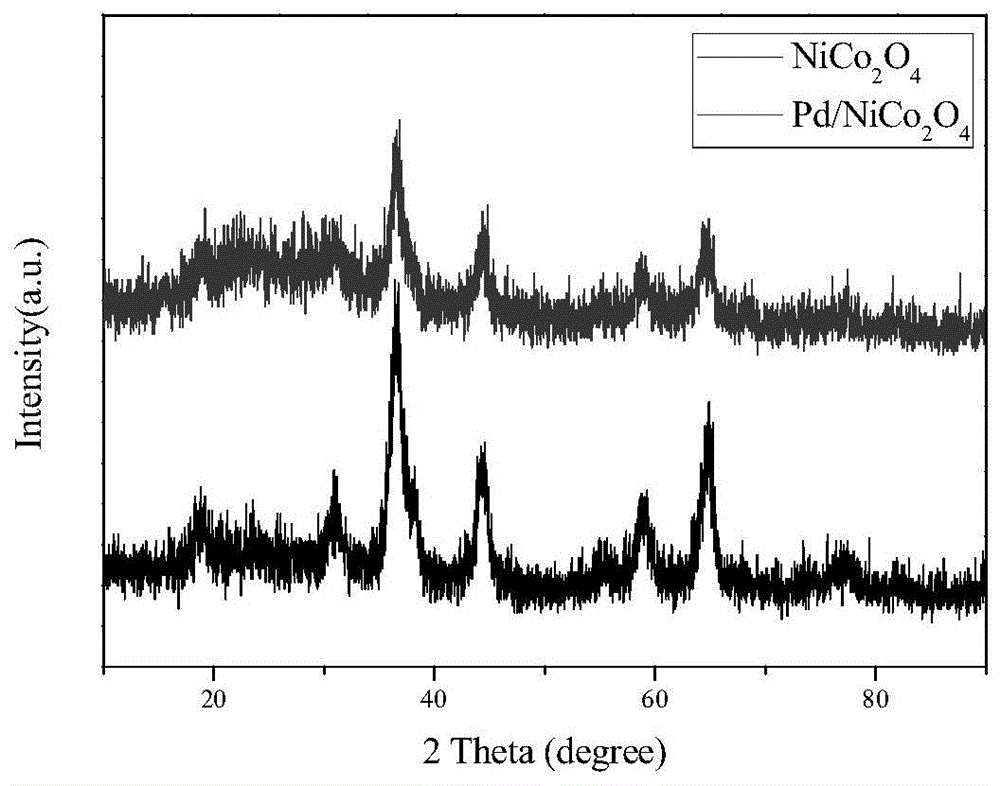
图1为本发明实施例1制备的nico2o4(下)以及pd/nico2o4(上)的x-射线衍射(xrd)图;
图2为本发明实施例1制备的催化剂的透射电子显微镜(tem)图。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合具体实施方式对本发明进一步详细说明。应当理解,此处所描述的具体实施方式仅仅用以解释本发明,但并不用于限定本发明。
本发明公开了一种用于α,β-不饱和醛/酮选择性加氢的催化剂,该催化剂用于定向催化α,β-不饱和醛/酮中的c=c键的加氢反应;催化剂包括活性组分钯以及多孔材料催化剂载体;活性组分钯与多孔材料催化剂载体的质量比为(0.1~10.0):100;多孔材料催化剂载体为尖晶石型结构化合物。
其中,催化剂中的活性组分钯以纳米粒子形式负载在多孔材料催化剂载体上,其粒径细小且均匀,并且高度分布在多孔材料催化剂载体上,从而能够使催化剂的催化活性高、稳定性高,催化c=c键的加氢反应选择性高、转化率高,且催化条件更为温和,能够在常压且较低的温度条件下进行定向加氢反应,并且催化加氢使用的溶剂更加绿色安全,易于回收处理,降低了投资成本、减少能耗,并且降低了对环境的不良影响。
作为一种可选实施方式,尖晶石型结构化合物为镍钴酸盐或镍钴酸盐复合物。尖晶石型结构化合物由多组分的组成,价态和电子结构在磁、光、电、催化领域受到广泛关注。特别是尖晶石型镍钴酸盐(nico2o4)因其组分多、表面积大、结构精确、吸附能力强等特点,已被广泛地应用于离子电池负极材料中。在本发明中通过研究发现,通过尖晶石型镍钴酸盐负载活性组分钯,由于尖晶石型镍钴酸盐的价态和电子结构,能够使活性组分钯在其作用下催化的选择性以及转化率更高,并且由于尖晶石型镍钴酸盐组分多、表面积大、结构精确、吸附能力强的特点,可以对底物具有一定的吸附、诱导作用,从而能够提高转化频率。进一步地,可以通过对镍钴酸盐进一步地复合,形成一定的复合物以针对特定的底物进行催化加氢。
可选地,活性组分钯与多孔材料催化剂载体的质量比为(0.5~1.5):100。例如,催化剂中活性组分钯与多孔材料催化剂载体的质量比可以是0.5:100、0.6:100、0.7:100、0.8:100、0.9:100、1.0:100、1.1:100、1.2:100、1.3:100、1.4:100、1.5:100。
本发明还提供了一种上述用于α,β-不饱和醛/酮选择性加氢的催化剂的制备方法,该制备方法包括以下步骤:
催化剂浸渍制备:将多孔材料催化剂载体与活性组分钯的金属盐溶液混合浸渍反应1h~24h,制得催化剂浸渍混合物;
还原处理:将催化剂浸渍混合物与还原剂溶液混合反应1h~3h,制得催化剂。
可选地,在还原处理步骤后,将制得的催化剂分别用水、无水乙醇洗涤后,于50℃~70℃真空干燥10h~14h后备用。
上述用于α,β-不饱和醛/酮选择性加氢的催化剂的制备方法,首先将活性组分钯以金属盐的形式浸渍于多孔材料催化剂载体上,由于多孔材料催化剂载体特殊的价态和电子结构,并且其组分多、表面积大、结构精确、吸附能力强的特性,能够使活性组分钯离子在多孔材料催化剂载体上高度分布,进一步通过还原剂进行还原钯离子,从而能够被还原为粒径细小的、纳米级的活性组分钯粒子,且其在多孔材料催化剂载体分布更为均匀,研究发现,这种钯粒子的粒径以及分布形态对于活性组分钯粒子发挥催化作用具有重要作用。
作为一种可选实施方式,作为多孔材料催化剂载体的尖晶石型结构化合物可以通过高温固相法、机械化学合成法、溶胶凝胶法、液相化学共沉淀法、喷雾热分解法、水热法、共沉淀热分解法中的任意一种方法制备而成。
可选地,当选用尖晶石型镍钴酸盐作为多孔材料催化剂载体时,尖晶石型镍钴酸盐的制备方法如下:
将可溶性镍盐、可溶性钴盐与乙酸钠或三水乙酸钠搅拌混合,直至生成紫红色溶液;
将生成的紫红色溶液与聚乙二醇混合置于150℃~250℃温度条件下反应12h~20h,反应生成的沉淀物经去离子水、无水乙醇洗涤后,于50℃~70℃温度条件下真空干燥10h~14h,制得镍钴酸盐前驱体;
将镍钴酸盐前驱体于300℃~800℃温度条件下煅烧1h~3h,制得尖晶石型钴镍酸盐。
上述尖晶石型钴镍酸盐制备方法,制备过程简单,环境友好,重现性好,且制得的尖晶石型钴镍酸盐呈现良好的多孔性特征,对活性组分钯具有良好的负载特性。
其中,可溶性镍盐可以是硝酸镍、六水硝酸镍、氯化镍、六水氯化镍、硫酸镍、七水硫酸镍中的任意一种。可溶性钴盐可以是硝酸钴、六水硝酸钴、氯化钴、六水氯化钴、硫酸钴、七水硫酸钴中的任意一种。聚乙二醇可以是聚乙二醇-200。
进一步地可选地,将镍钴酸盐前驱体于300℃~800℃温度条件下煅烧1h~3h,可以是将镍钴酸盐前驱体置于马弗炉中,程序升温至300℃~800℃进行煅烧。例如,可以以2.0℃/min~4.0℃/min的升温速率升温至300℃~800℃进行煅烧。优选地,镍钴酸盐前驱体的煅烧温度为300℃~380℃。镍钴酸盐由于热不稳定性质,若温度过高则会部分分解生成一些简单氧化物和富含钴的尖晶石相,使其催化活性降低,经过研究发现,当镍钴酸盐前驱体的煅烧温度为300℃~380℃时,能够明显避免上述分解问题产生。进一步地,当煅烧温度为300℃~380℃时,生成的尖晶石型镍钴酸盐的粒径为25nm~30nm,其粒度均一,更为便于其负载活性组分钯以及便于制备的催化剂的使用。
作为一种可选实施方式,活性组分钯的金属盐为四氯钯酸钾、氯化钯、二氯四氨合钯中的任意一种。可选地,活性组分钯的金属盐溶液的摩尔浓度为0.01mol/l~0.10mol/l,优选地,活性组分钯的金属盐溶液的摩尔浓度为0.02mol/l~0.05mol/l。
优选地,活性组分钯的金属盐为四氯钯酸钾(k2pdcl4),四氯钯酸钾易于溶解于水,且溶解后不需要进一步调节ph,制备方法更为简便且以四氯钯酸钾作为钯源时,制得的催化剂的转化率以及催化选择性较高。而当选用氯化钯时,为了提高氯化钯在水中的溶解度,可以加入适量的盐酸。例如,活性组分钯的金属盐溶液可以是0.02mol/l的四氯钯酸钾溶液。
作为一种可选实施方式,上述催化剂的制备方法中的还原剂为硼氢化钠,在所述还原步骤中,活性组分钯的金属盐与硼氢化钠的摩尔比为1:(1~9)。
利用上述实施例制备的用于α,β-不饱和醛/酮选择性加氢的催化剂进行催化反应时,无需高温高压,催化反应的条件更为温和,且无需使用高污染性的反应试剂。可选地,本发明制备的催化剂催化α,β-不饱和醛/酮选择加氢的催化方法包括以下步骤:
将α,β-不饱和醛/酮底物、催化剂以及溶剂于常压下通入氢气反应,反应温度为30℃~60℃。
可选地,催化反应后的催化剂分别用水,无水乙醇洗涤后,于50℃~70℃真空干燥10h~14h后可复活重复加以利用。
可选地,选用的溶剂可以是无水乙醇。无水乙醇无论是试剂成本还是后处理成本都更为低廉,且对环境无污染,是一种绿色试剂。
优选地,催化剂的用量为α,β-不饱和醛/酮底物质量的1.0%~1.5%。
可选地,α,β-不饱和醛/酮底物包括肉桂醛、苄叉丙酮、2-环己烯-1-酮、2-甲基-2-戊烯醛、巴豆醛、3-庚烯-2-酮。
催化剂制备实施例1
称取一定量的硝酸镍、硝酸钴和60mmol的乙酸钠·三水混合置100ml的聚四氟乙烯的反应釜中,搅拌直到紫红色溶液形成。加入40ml聚乙二醇-200,磁力搅拌1h混合,将混合液置于200℃条件下反应16h。当反应釜降至室温后,将反应釜中沉淀物抽滤,用去离子水和无水乙醇洗涤几次,将所得到的产物置于真空干燥箱中于60℃干燥12h,得到蓝绿色固体粉末状的镍钴酸盐前驱体。将该蓝绿色固体粉末状的镍钴酸盐前驱体研磨后置于马弗炉中,于350℃温度条件下煅烧处理2h,得到黑色固体粉末状的多孔材料催化剂载体——尖晶石型钴镍酸盐,其xrd图如图1所示。
称取198mg上述尖晶石型钴镍酸盐,加入80ml水中搅拌分散或超声分散1h,滴加0.94ml的0.02mol/l的k2pdcl4溶液,搅拌浸渍反应12h。用滴管慢慢滴加含有6.27×10-3molnabh4的nabh4溶液,反应2h后,经抽滤、去离子水洗涤、60℃真空干燥12h,得到呈黑色粉末状的nico2o4负载pd的催化剂,记为pd/nico2o4催化剂a1。该pd/nico2o4催化剂a1的xrd图和tem图分别如图1和图2所示。
催化剂制备实施例2
称取一定量的硝酸镍、硝酸钴和60mmol的乙酸钠·三水混合置100ml的聚四氟乙烯的反应釜中,搅拌直到紫红色溶液形成。加入40ml聚乙二醇-200,磁力搅拌1h混合,将混合液置于200℃条件下反应16h。当反应釜降至室温后,将反应釜中沉淀物抽滤,用去离子水和无水乙醇洗涤几次,将所得到的产物置于真空干燥箱中于60℃干燥12h,得到蓝绿色固体粉末状的镍钴酸盐前驱体。将该蓝绿色固体粉末状的镍钴酸盐前驱体研磨后置于马弗炉中,于350℃温度条件下煅烧处理2h,得到黑色固体粉末状的多孔材料催化剂载体——尖晶石型钴镍酸盐。
称取198mg上述尖晶石型钴镍酸盐,加入80ml水中搅拌分散或超声分散1h,滴加0.47ml的0.02mol/l的k2pdcl4溶液,搅拌浸渍反应12h。用滴管慢慢滴加含有6.27×10-3molnabh4的nabh4溶液,反应2h后,经抽滤、去离子水洗涤、60℃真空干燥12h,得到呈黑色粉末状的nico2o4负载pd的催化剂,记为pd/nico2o4催化剂a2。
催化剂制备实施例3
称取一定量的硝酸镍、硝酸钴和60mmol的乙酸钠·三水混合置100ml的聚四氟乙烯的反应釜中,搅拌直到紫红色溶液形成。加入40ml聚乙二醇-200,磁力搅拌1h混合,将混合液置于200℃条件下反应16h。当反应釜降至室温后,将反应釜中沉淀物抽滤,用去离子水和无水乙醇洗涤几次,将所得到的产物置于真空干燥箱中于60℃干燥12h,得到蓝绿色固体粉末状的镍钴酸盐前驱体。将该蓝绿色固体粉末状的镍钴酸盐前驱体研磨后置于马弗炉中,于350℃温度条件下煅烧处理2h,得到黑色固体粉末状的多孔材料催化剂载体——尖晶石型钴镍酸盐。
称取198mg上述尖晶石型钴镍酸盐,加入80ml水中搅拌分散或超声分散1h,滴加1.41ml的0.02mol/l的k2pdcl4溶液,搅拌浸渍反应12h。用滴管慢慢滴加含有6.27×10-3molnabh4的nabh4溶液,反应2h后,经抽滤、去离子水洗涤、60℃真空干燥12h,得到呈黑色粉末状的nico2o4负载pd的催化剂,记为pd/nico2o4催化剂a3。
催化剂制备实施例4
称取一定量的硝酸镍、硝酸钴和60mmol的乙酸钠·三水混合置100ml的聚四氟乙烯的反应釜中,搅拌直到紫红色溶液形成。加入40ml聚乙二醇-200,磁力搅拌1h混合,将混合液置于200℃条件下反应16h。当反应釜降至室温后,将反应釜中沉淀物抽滤,用去离子水和无水乙醇洗涤几次,将所得到的产物置于真空干燥箱中于60℃干燥12h,得到蓝绿色固体粉末状的镍钴酸盐前驱体。将该蓝绿色固体粉末状的镍钴酸盐前驱体研磨后置于马弗炉中,于350℃温度条件下煅烧处理2h,得到黑色固体粉末状的多孔材料催化剂载体——尖晶石型钴镍酸盐。
称取198mg上述尖晶石型钴镍酸盐,加入80ml水中搅拌分散或超声分散1h,滴加0.09ml的0.02mol/l的k2pdcl4溶液,搅拌浸渍反应12h。用滴管慢慢滴加含有6.27×10-3molnabh4的nabh4溶液,反应2h后,经抽滤、去离子水洗涤、60℃真空干燥12h,得到呈黑色粉末状的nico2o4负载pd的催化剂,记为pd/nico2o4催化剂a4。
催化剂制备实施例5
称取一定量的硝酸镍、硝酸钴和60mmol的乙酸钠·三水混合置100ml的聚四氟乙烯的反应釜中,搅拌直到紫红色溶液形成。加入40ml聚乙二醇-200,磁力搅拌1h混合,将混合液置于200℃条件下反应24h。当反应釜降至室温后,将反应釜中沉淀物抽滤,用去离子水和无水乙醇洗涤几次,将所得到的产物置于真空干燥箱中于60℃干燥12h,得到蓝绿色固体粉末状的镍钴酸盐前驱体。将该蓝绿色固体粉末状的镍钴酸盐前驱体研磨后置于马弗炉中,于350℃温度条件下煅烧处理2h,得到黑色固体粉末状的多孔材料催化剂载体——尖晶石型钴镍酸盐。
称取198mg上述尖晶石型钴镍酸盐,加入80ml水中搅拌分散或超声分散1h,滴加9.40ml的0.02mol/l的k2pdcl4溶液,搅拌浸渍反应1h。用滴管慢慢滴加含有6.27×10-3molnabh4的nabh4溶液,反应2h后,经抽滤、去离子水洗涤、60℃真空干燥12h,得到呈黑色粉末状的nico2o4负载pd的催化剂,记为pd/nico2o4催化剂a5。
催化剂制备实施例6
称取一定量的硝酸镍、硝酸钴和60mmol的乙酸钠·三水混合置100ml的聚四氟乙烯的反应釜中,搅拌直到紫红色溶液形成。加入40ml聚乙二醇-200,磁力搅拌1h混合,将混合液置于200℃条件下反应16h。当反应釜降至室温后,将反应釜中沉淀物抽滤,用去离子水和无水乙醇洗涤几次,将所得到的产物置于真空干燥箱中于60℃干燥12h,得到蓝绿色固体粉末状的镍钴酸盐前驱体。将该蓝绿色固体粉末状的镍钴酸盐前驱体研磨后置于马弗炉中,于350℃温度条件下煅烧处理2h,得到黑色固体粉末状的多孔材料催化剂载体——尖晶石型钴镍酸盐。
称取198mg上述尖晶石型钴镍酸盐,加入80ml水中搅拌分散或超声分散1h,滴加0.94ml的0.02mol/l的k2pdcl4溶液,搅拌浸渍反应12h。用滴管慢慢滴加含有4.18×10-3molnabh4的nabh4溶液,反应2h后,经抽滤、去离子水洗涤、60℃真空干燥12h,得到呈黑色粉末状的nico2o4负载pd的催化剂,记为pd/nico2o4催化剂a6。
催化剂制备实施例7
称取一定量的硝酸镍、硝酸钴和60mmol的乙酸钠·三水混合置100ml的聚四氟乙烯的反应釜中,搅拌直到紫红色溶液形成。加入40ml聚乙二醇-200,磁力搅拌1h混合,将混合液置于200℃条件下反应16h。当反应釜降至室温后,将反应釜中沉淀物抽滤,用去离子水和无水乙醇洗涤几次,将所得到的产物置于真空干燥箱中于60℃干燥12h,得到蓝绿色固体粉末状的镍钴酸盐前驱体。将该蓝绿色固体粉末状的镍钴酸盐前驱体研磨后置于马弗炉中,于350℃温度条件下煅烧处理2h,得到黑色固体粉末状的多孔材料催化剂载体——尖晶石型钴镍酸盐。
称取198mg上述尖晶石型钴镍酸盐,加入80ml水中搅拌分散或超声分散1h,滴加0.94ml的0.02mol/l的k2pdcl4溶液,搅拌浸渍反应12h。用滴管慢慢滴加含有8.36×10-3molnabh4的nabh4溶液,反应2h后,经抽滤、去离子水洗涤、60℃真空干燥12h,得到呈黑色粉末状的nico2o4负载pd的催化剂,记为pd/nico2o4催化剂a7。
催化剂制备对比例1
采用实施例1制备的尖晶石型钴镍酸盐作为本对比例的多孔材料催化剂载体。
将0.38ml的0.05mol/l的pdcl2·2hcl用去离子水稀释至浓度为10-3mol/l并快速搅拌,然用滴管缓慢滴加1.0mol/l的nabh4溶液,氯酸钯与硼氢化钠的摩尔比为3:1,然后用1.0mol/lkoh调节混合液的ph至9~10,磁力搅拌还原反应2h。加入200mg实施例1制备的尖晶石型钴镍酸盐作为多孔材料催化剂载体,搅拌浸渍反应12h,将沉淀物经抽滤、去离子水洗涤、60℃真空干燥12h,得到呈黑色粉末状的nico2o4负载pd的催化剂,记为pd/nico2o4催化剂b。
催化剂制备对比例2
称取一定量的硝酸钴和60mmol的乙酸钠·三水混合置100ml的聚四氟乙烯的反应釜中,搅拌直到紫红色溶液形成。加入40ml聚乙二醇-200,磁力搅拌1h混合,将混合液置于200℃条件下反应16h。当反应釜降至室温后,将反应釜中沉淀物抽滤,用去离子水和无水乙醇洗涤几次,将所得到的产物置于真空干燥箱中于60℃干燥12h,得到蓝绿色固体粉末状的镍钴酸盐前驱体。将该蓝绿色固体粉末状的镍钴酸盐前驱体研磨后置于马弗炉中,于350℃温度条件下煅烧处理2h,得到co3o4载体。
称取198mg上述co3o4载体,加入80ml水中搅拌分散或超声分散1h,滴加0.94ml的0.02mol/l的k2pdcl4溶液,搅拌浸渍反应12h。用滴管慢慢滴加含有6.27×10-3molnabh4的nabh4溶液,反应2h后,经抽滤、去离子水洗涤、60℃真空干燥12h,得到co3o4负载pd的催化剂,记为pd/co3o4催化剂c。
催化剂制备对比例3
称取一定量的硝酸镍和60mmol的乙酸钠·三水混合置100ml的聚四氟乙烯的反应釜中,搅拌直到紫红色溶液形成。加入40ml聚乙二醇-200,磁力搅拌1h混合,将混合液置于200℃条件下反应16h。当反应釜降至室温后,将反应釜中沉淀物抽滤,用去离子水和无水乙醇洗涤几次,将所得到的产物置于真空干燥箱中于60℃干燥12h,得到蓝绿色固体粉末状的镍钴酸盐前驱体。将该蓝绿色固体粉末状的镍钴酸盐前驱体研磨后置于马弗炉中,于350℃温度条件下煅烧处理2h,得到nio2载体。
称取198mg上述nio2载体,加入80ml水中搅拌分散或超声分散1h,滴加0.94ml的0.02mol/l的k2pdcl4溶液,搅拌浸渍反应12h。用滴管慢慢滴加含有6.27×10-3molnabh4的nabh4溶液,反应2h后,经抽滤、去离子水洗涤、60℃真空干燥12h,得到nio2负载pd的催化剂,记为pd/nio2催化剂d。
催化反应实施例1
以50ml的圆底烧瓶作为本实施例的反应容器,催化反应前,烧瓶内气体经氢气20ml/min置换5分钟。
催化反应实验条件为:50mg的pd/nico2o4催化剂a1、20ml的无水乙醇、0.2ml肉桂醛(cal)置于反应容器内,常压下通h2,h2的气流量为20ml/min,控制催化反应温度为30℃~60℃,以苄醇为内标物,反应液过滤后取上清液通过气相色谱仪进行分析,分析结果如表1所示。
反应后的pd/nico2o4催化剂a1分别用纯水清洗3次,无水乙醇清洗3次,然后将该pd/nico2o4催化剂a1在真空干燥箱中于60℃干燥12h,经真空干燥后该催化剂可重复利用。
催化反应对比例1
以50ml的圆底烧瓶作为本实施例的反应容器,催化反应前,烧瓶内气体经氢气20ml/min置换5分钟。
催化反应实验条件为:50mg的pd/nico2o4催化剂b、20ml的无水乙醇、0.2ml肉桂醛(cal)置于反应容器内,常压下通h2,h2的气流量为20ml/min,控制催化反应温度为30℃~60℃,以苄醇为内标物,反应液过滤后取上清液通过气相色谱仪进行分析,分析结果如表1所示。
催化反应实施例2
以50ml的圆底烧瓶作为本实施例的反应容器,催化反应前,烧瓶内气体经氢气20ml/min置换5分钟。
催化反应实验条件为:50mg的pd/co3o4催化剂c、20ml的无水乙醇、0.2ml肉桂醛(cal)置于反应容器内,常压下通h2,h2的气流量为20ml/min,控制催化反应温度为30℃~60℃,以苄醇为内标物,反应液过滤后取上清液通过气相色谱仪进行分析,分析结果如表1所示。
催化反应对比例3
以50ml的圆底烧瓶作为本实施例的反应容器,催化反应前,烧瓶内气体经氢气20ml/min置换5分钟。
催化反应实验条件为:50mg的pd/nio2催化剂d、20ml的无水乙醇、0.2ml肉桂醛(cal)置于反应容器内,常压下通h2,h2的气流量为20ml/min,控制催化反应温度为30℃~60℃,以苄醇为内标物,反应液过滤后取上清液通过气相色谱仪进行分析,分析结果如表1所示。
表1不同催化剂催化效果分析结果
从表1可以看出,本发明催化剂制备实施例1制备的pd/nico2o4催化剂a1催化肉桂醛加氢反应时,c=c键加氢反应选择性达到99.9%,转化率达到99.9%,具有优良的选择加氢效果。而催化剂制备对比例1制备的pd/nico2o4催化剂b虽然理论上具备与pd/nico2o4催化剂a1相同的结构,但是由于制备条件不同,先还原反应然后再浸渍反应,其催化效果较pd/nico2o4催化剂a1差很多,催化肉桂醛c=c键加氢选择性以及转化率远低于pd/nico2o4催化剂a1。催化剂制备对比例2制备的pd/co3o4催化剂c催化肉桂醛c=c键加氢转化率和选择性均较pd/nico2o4催化剂a1低。催化剂制备对比例3制备的pd/nio2催化剂d催化肉桂醛c=c键加氢的选择性虽然能够达到100%,但是其转化率远低于pd/nico2o4催化剂a1,难以工业化应用。
进一步地,为了说明本发明制备的pd/nico2o4催化剂a1对c=c和c=o双键的催化加氢选择性,采用与催化反应实施例1相同的反应条件,催化底物分别为苄叉丙酮、2-环己烯-1-酮、2-甲基-2-戊烯醛、巴豆醛、3-庚烯-2-酮和2-甲基丙烯醛,反应液过滤后取上清液通过气相色谱仪进行分析,分析结果如表2所示。
表2不同α,β-不饱和醛/酮底物的催化效果分析结果
从表2可以看出,本发明制备的pd/nico2o4催化剂a的除对2-甲基丙烯醛的加氢反无催化作用外,对其他α,β-不饱和醛/酮均具有选择催化的效果,并且对苄叉丙酮、2-环己烯-1-酮、2-甲基-2-戊烯醛、巴豆醛、3-庚烯-2-酮的c=c键催化加氢转化率以及选择性均达到99.9%以上,并且其转化频率也极高。虽然对异佛尔酮的c=c键催化加氢转化率和转化频率不高,但是其选择性达到了100%,然而申请人认为基于本申请的发明构思可通过对pd/nico2o4催化剂a进一步改性研究以实现其催化效果的改进提高。
进一步地,为了说明本发明制备的pd/nico2o4催化剂对c=c和c=o双键的催化加氢选择性,采用与催化反应实施例1相同的反应条件,催化剂分别为pd/nico2o4催化剂a2、pd/nico2o4催化剂a3、pd/nico2o4催化剂a4、pd/nico2o4催化剂a5、pd/nico2o4催化剂a6、pd/nico2o4催化剂a7,用于催化肉桂醛的c=c键加氢,反应液过滤后取上清液通过气相色谱仪进行分析,分析结果如表3所示。
表3pd/nico2o4催化剂a1-a7的催化效果分析结果
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
- 还没有人留言评论。精彩留言会获得点赞!