三元水滑石吸附剂及其制备方法和应用与流程
本发明涉及水滑石及其制备和应用。
背景技术:
人类沿海地区经济发展及海上活动的日益增加,导致近岸环境质量逐年下降,特别是磷等营养盐的浓度不断增加引起的海水富营养化,对海洋环境以及人类的健康造成了威胁。为了应对上述问题,寻求一种对水中磷酸根具有快速有效吸附能力的材料是吸附法去除水溶液以及海水中磷酸根的关键。
类水滑石材料具有良好的物理化学性能,层板元素可调控、层间阴离子可交换、稳定性高等特性使其在电催化、电容器电极材料和吸附重金属等方面被广泛应用。但在吸附磷酸根,特别是海水中磷酸根的研究相对较少,作为一种特殊的层状化合物,类水滑石材料各向异性的性质使得其易于形成离子吸附和运输的二维渗透通道。中国科学技术大学硕士论文《天然纤维素和水滑石的改性及其对重金属和磷酸根的吸咐研究》一文中,以镁铝水滑石为基底,过[feedta]-络合阴离子对水滑石进行修饰改性,在镁铝水滑石上负载水合氧化铁得到一种复合材料,并运用在磷酸根的吸附去除实验中。改性后的吸附剂对磷酸根的吸附能力提高,但是由于制备过程采用[feedta]-络合阴离子,使得吸附过程中除了阴离子交换还包括络合过程。由于络合剂的生物降解能力低,因此这种吸附机制虽然会增大吸附量,但也会带来污染的危险。同时由于有机离子的存在,吸咐后再生困难。
技术实现要素:
本发明是要解决现有的水滑石类吸附剂的易产生污染、再生困难的技术问题,而提供一种三元水滑石吸附剂及其制备方法和应用。
本发明的三元水滑石吸附剂,其化学式为mg6mzr(oh)16co3﹒4h2o。其中m为二价或三价金属元素;用mgmzr-ldh表示。
上述的三元水滑石吸附剂的制备方法,按以下步骤进行:
一、按mg(no3)2·6h2o、二价或三价金属硝酸盐、锆盐的摩尔比6:(1~2):(1~2),将mg(no3)2·6h2o、二价或三价金属硝酸盐、锆盐加入水中溶解,得到混合盐溶液a;
二、按氢氧化钠与碳酸钠的摩尔比为(2~3):1,将氢氧化钠与碳酸钠加入水中溶解,得到混合溶液b;
三、在搅拌条件下,向步骤一得到的混合盐溶液a中滴加混合溶液b,滴加过程中控制整个溶液ph值在8~10之间,得到混合溶液c;
四、将混合溶液c在15℃~80℃的条件下搅拌8~24h,然后离心分离,将固相物洗涤干净,再经干燥、研磨,得到三元水滑石吸附剂;该三元水滑石吸附剂用mgmzr-ldh表示。
本发明的三元水滑石吸附剂,其化学式还可以是mg6mzr(oh)18,其中m为二价或三价金属元素。用mgmzr-ldo表示。
上述的三元水滑石吸附剂的制备方法,按以下步骤进行:
将mg6mzr(oh)16co3﹒4h2o在温度为130~300℃的马弗炉中煅烧3~5h,得到三元水滑石吸附剂,该三元水滑石吸附剂用mgmzr-ldo表示。
上述的三元水滑石吸附剂的应用,是将它用作吸附剂、催化剂或电极材料。
其中利用三元水滑石吸附剂吸附去除海水中po43-的方法,按以下步骤进行:
将三元水滑石吸附剂加入到ph值为7~8的含po43-的海水中,振荡后,离心分离,完成含po43-的海水的处理。
本发明采用来源广泛的混合盐为原料,引入锆元素,通过简单的共沉淀法制备三元水滑石吸附剂mgmzr-ldh(m=mn或al),还可以通过煅烧得到焙烧态的三元水滑石吸附剂mgmzr-ldo(m=mn或al)。本发明利用水滑石层板的元素的可调控性,引入高价态阳离子使得层板正电荷过剩,从而使得其具备层间阴离子更容易被交换;利用水滑石材料的记忆效应,化合物通过煅烧、层板的再组装,使其具有更好的热稳定性和高的分散性等特质,这些都利于吸附带负电荷的磷酸根离子。用本发明的三元水滑石吸附剂处理含磷废水,mgmnzr-ldh、mgmnzr-ldo、mgalzr-ldh和mgalzr-ldo的吸附量均超过10mg/g,表现出了较好的吸附性能。同时吸附平衡时间短,约100min以内就能达到吸咐平衡,大大降低了处理含磷废水处理的时间成本,吸附后的产物可以通过离心分离的方法与水体进行分离回收,有利于环境保护,并且吸附后的吸附剂可以通过氢氧化钠、碳酸钠或其混合溶液进行脱吸附处理进行再生。由于元素锆的引入,增加了吸附剂的结构稳定性,在竞争离子较多的海水中能够选择性地吸咐po43-,并且在多次循环再生后使用仍然具有较好的吸附量,同时不产生二次污染。
本发明的三元水滑石吸附剂可用于电池领域、污染海水处理领域。
附图说明
图1是实施例1制备的三元水滑石的结构示意图;
图2是实施例1制备的三元水滑石的tg-dta图;
图3是实施例1制备的三元水滑石及其焙烧物的xrd谱图;
图4是实施例1制备的三元水滑石mgmnzr-ldh的sem图;
图5是实施例2制备的三元水滑石mgmnzr-ldo130的sem图;
图6是实施例3制备的三元水滑石mgmnzr-ldo500的sem图;
图7是实施例1制备的三元水滑石吸附剂mgmnzr-ldh、实施例2制备的mgmnzr-ldo130和实施例3制备的mgmnzr-ldo500的比表面积对比图;
图8是实施例1制备的三元水滑石吸附剂mgmnzr-ldh、实施例2制备的mgmnzr-ldo130和实施例3制备的mgmnzr-ldo500吸附磷酸根的吸咐曲线;
图9是实施例1中mgmnzr-ldh三元水滑石吸附磷酸根的动力学模拟曲线;
图10是实施例1中mgmnzr-ldh吸附海水中磷酸根后的epma图;
图11是实施例4制备的三元水滑石mgalzr-ldh的tg-dta图;
图12是实施例4制备的三元水滑石吸附剂mgalzr-ldh、实施例5制备的mgalzr-ldo130和实施例6制备的mgalzr-ldo500的xrd谱图;
图13是实施例4制备的三元水滑石mgalzr-ldh的sem图;
图14是实施例5制备的三元水滑石mgalzr-ldo130的sem图;
图15是实施例6制备的三元水滑石mgalzr-ldo500的sem图;
图16是实施例4制备的三元水滑石吸附剂mgalzr-ldh、实施例5制备的mgalzr-ldo130和实施例6制备的mgalzr-ldo500的比表面积、孔容积和孔径对比图;
图17是实施例4制备的三元水滑石吸附剂mgalzr-ldh、实施例5制备的mgalzr-ldo130和实施例6制备的mgalzr-ldo500吸附磷酸根的吸咐曲线;
图18是实施例4中mgalzr-ldh三元水滑石吸附磷酸根的动力学模拟曲线;
图19是实施例4中mgalzr-ldh吸附海水中磷酸根后的epma图。
具体实施方式
具体实施方式一:本实施方式的三元水滑石吸附剂,其化学式为mg6mzr(oh)16co3﹒4h2o。其中m为二价或三价金属元素。用mgmzr-ldh表示。
具体实施方式二:本实施方式与具体实施方式一不同的是m为mn或al;其他与具体实施方式一相同。
具体实施方式三:具体实施方式一所述的三元水滑石吸附剂的制备方法,按以下步骤进行:
一、按mg(no3)2·6h2o、二价或三价金属硝酸盐、锆盐的摩尔比6:(1~2):(1~2),将mg(no3)2·6h2o、二价或三价金属硝酸盐、锆盐加入水中溶解,得到混合盐溶液a;
二、按氢氧化钠与碳酸钠的摩尔比为(2~3):1,将氢氧化钠与碳酸钠加入水中溶解,得到混合溶液b;
三、在搅拌条件下,向步骤一得到的混合盐溶液a中滴加混合溶液b,滴加过程中控制整个溶液ph值在8~10之间,得到混合溶液c;
四、将混合溶液c在15℃~80℃的条件下搅拌8~24h,然后离心分离,将固相物洗涤干净,再经干燥、研磨,得到三元水滑石吸附剂;该三元水滑石吸附剂用mgmzr-ldh表示。
具体实施方式四:本实施方式与具体实施方式三不同的是步骤一中所述的二价或三价金属硝酸盐为mn(no3)2·4h2o或al(no3)3·9h2o;其它与具体实施方式三相同。
具体实施方式五:本实施方式与具体实施方式三或四不同的是步骤一中所述的锆盐为zr(no3)4·5h2o或zrcl2o·8h2o。其它与具体实施方式三或四相同。
具体实施方式六:本实施方式与具体实施方式三至五之一不同的是步骤一中的mn(no3)2·9h2o与步骤二中氢氧化钠的摩尔比为1:(0.3~0.5)。其它与具体实施方式三至五之一相同。
具体实施方式七:本发明实施方式的三元水滑石吸附剂,其化学式是mg6mzr(oh)18,其中m为二价或三价金属元素。用mgmzr-ldo表示。
具体实施方式八:本实施方式与具体实施方式七不同的是m为mn或al;其它与具体实施方式七相同。
具体实施方式九:具体实施方式八所述的三元水滑石吸附剂的制备方法,按以下步骤进行:将mg6mzr(oh)16co3﹒4h2o在温度为130~300℃的马弗炉中煅烧3~5h,得到三元水滑石吸附剂。该三元水滑石吸附剂用mgmzr-ldo表示。
具体实施方式十:具体实施方式一或八所述的三元水滑石吸附剂的应用,是将它用作吸附剂、催化剂或电极材料。
具体实施方式十一:利用三元水滑石吸附剂吸附去除海水中po43-的方法,按以下步骤进行:将三元水滑石吸附剂加入到ph值为7~8的含po43-的海水中,振荡后,将三元水滑石吸附剂分离出来,完成含po43-的海水的处理。
具体实施方式十二:本实施方式与具体实施方式十一不同的是三元水滑石吸附剂的加入量为0.1~0.15g/l。其它与具体实施方式十一相同。
具体实施方式十三:本实施方式与具体实施方式十一或十二不同的是振荡时间为10min~120min。其它与具体实施方式十一或十二相同。
用下面的实施例验证本发明的有益效果:
实施例1:本实施例1的三元水滑石吸附剂mgmnzr-ldh的制备方法,按以下步骤进行:
一、按mg(no3)2·6h2o、mn(no3)2·4h2o、zrcl2o·8h2o摩尔比6:1:1,将15.38gmg(no3)2·6h2o、2.51gmn(no3)2·4h2o、3.22gzrcl2o·8h2o加入100ml水中溶解,得到混合盐溶液a;
二、按氢氧化钠与碳酸钠的摩尔比为2:1,将1.2g氢氧化钠与1.59g碳酸钠加入150ml水中溶解,得到混合溶液b;
三、在搅拌条件下,向步骤一得到的混合盐溶液a中滴加混合溶液b,滴加过程中控制整个溶液ph值在8~9之间,得到混合溶液c;
四、将混合溶液c在80℃的条件下搅拌12h,然后离心分离,将固相物用去离子水洗涤干净,再经在60℃条件下干燥48h、研磨,得到三元水滑石吸附剂mgmnzr-ldh。
实施例2:本实施例1的三元水滑石吸附剂mgmnzr-ldo的制备方法,按以下步骤进行:
将实施例1制备的三元水滑石吸附剂mgmnzr-ldh放在温度为130℃的马弗炉中煅烧4h,得到三元水滑石吸附剂,该三元水滑石吸附剂用mgmnzr-ldo130表示。
实施例3:本实施例是实施例2的对比试验,本实施例与实施例2不同的是煅烧温度为500℃,其它与实施例2相同,得到作为对比的三元水滑石,用mgmnzr-ldo500表示。
实施例1所制备的三元水滑石吸附剂mgmnzr-ldh的层状结构示意图如图1所示,水滑石类化合物是有带正电荷的主体层板和层间阴离子通过共价键的相互作用组装成的化合物,具有典型的层状结构。
实施例1制备的三元水滑石吸附剂mgmnzr-ldh的热失重变化tg-dta如图2所示:通过观察图2中100℃和370℃左右的两个吸热峰,低温部分的吸热峰推断是产物失去游离水,说明产物仍然维持层状结构,而高温测的吸热峰则是层间的水分子以及碳酸跟离子分解,层间只留下了羟基引起的。
实施例1制备的三元水滑石吸附剂mgmnzr-ldh、实施例2制备的mgmnzr-ldo130和实施例3制备的mgmnzr-ldo500的xrd谱图如图3所示,从图3可以看出,mgmnzr-ldh、mgmnzr-ldo130和mgmnzr-ldo500都具有明显的层状结构,但是衍射峰的强度不同,这是由于煅烧处理使层间的水分子和碳酸根离子消失或转化为半径较小的羟基,有一部分不定型层形成的原因。在煅烧500℃条件下制备的mgmnzr-ldo500的衍射峰的位置有了很大偏离,说明层间的羟基消失,水滑石的层结构已经损坏。
实施例1制备的三元水滑石吸附剂mgmnzr-ldh的扫描电镜照片如图4所示,实施例2制备的mgmnzr-ldo130的扫描电镜照片如图5所示,实施例3制备的mgmnzr-ldo500的扫描电镜照片如图6所示,从图4~图6比较可以看出随着煅烧温度的升高,由于层状结构的坍塌,表面的凹凸程度变大,并且表明颗粒状增加。
实施例1制备的三元水滑石吸附剂mgmnzr-ldh、实施例2制备的mgmnzr-ldo130和实施例3制备的mgmnzr-ldo500的比表面积对比如图7所示,从图7可以看出,三元水滑石吸附剂mgmnzr-ldh的比表面积为168m2·g-1,孔容积是19.8×10-3cm3·g-1,孔径为10.9nm。mgmnzr-ldo130的比表面积为11.3m2·g-1,孔容积是25.1cm3·g-1,孔径为8.89nm。由于在吸附过程较大的比表面积中能够容纳更多的吸附质,另外吸附材料的孔径与磷酸跟离子的大小匹配程度也与吸附量有关,一般是越大吸附越容易。而mgmnzr-ldo500由于煅烧温度过高,层状结构的坍塌,水滑石的比表面积变小孔径变大,不利于吸咐。
将实施例1制备的三元水滑石吸附剂mgmnzr-ldh、实施例2制备的mgmnzr-ldo130和实施例3制备的mgmnzr-ldo500应用于吸附海水中的po43-,具体按以下步骤进行:
准确称取0.1g/lmgmnzr-ldh、mgmnzr-ldo130或mgmnzr-ldo500加入到ph值为8的含po43-的海水中,其中海水中po43-的浓度为5ppm;将其放入恒温震荡箱中,震荡120min,期间取上层清液,并用磷钼蓝分光光度法测上层清液中磷酸根的浓度,计算吸附量。mgmnzr-ldh、mgmnzr-ldo130或mgmnzr-ldo500的吸附量如图8所示。从图8可以看出,mgmnzr-ldh、mgmnzr-ldo130吸附磷酸根的效果较好,100min以内就能达到吸咐平衡,mgmnzr-ldh的平衡吸咐量达到15.8mg·g-1,mgmnzr-ldo130的平衡吸咐量达到13.9mg·g-1,而mgmnzr-ldo500因为层状结构的坍塌,吸附量较低。
为了进一步明确吸附过程中的机理,通过拟一阶和拟二阶动力学方程拟合,结果表明三元水滑石mgmnzr-ldh对磷酸根的吸附过程更符合拟二阶动力学模型,说明吸附过程是受拟二次方程支配的,如图9所示。证明吸附过程发生在不止发生在吸附剂表面。类似地,mgmnzr-ldo130对磷酸根的吸附过程也符合拟二阶动力学模型。
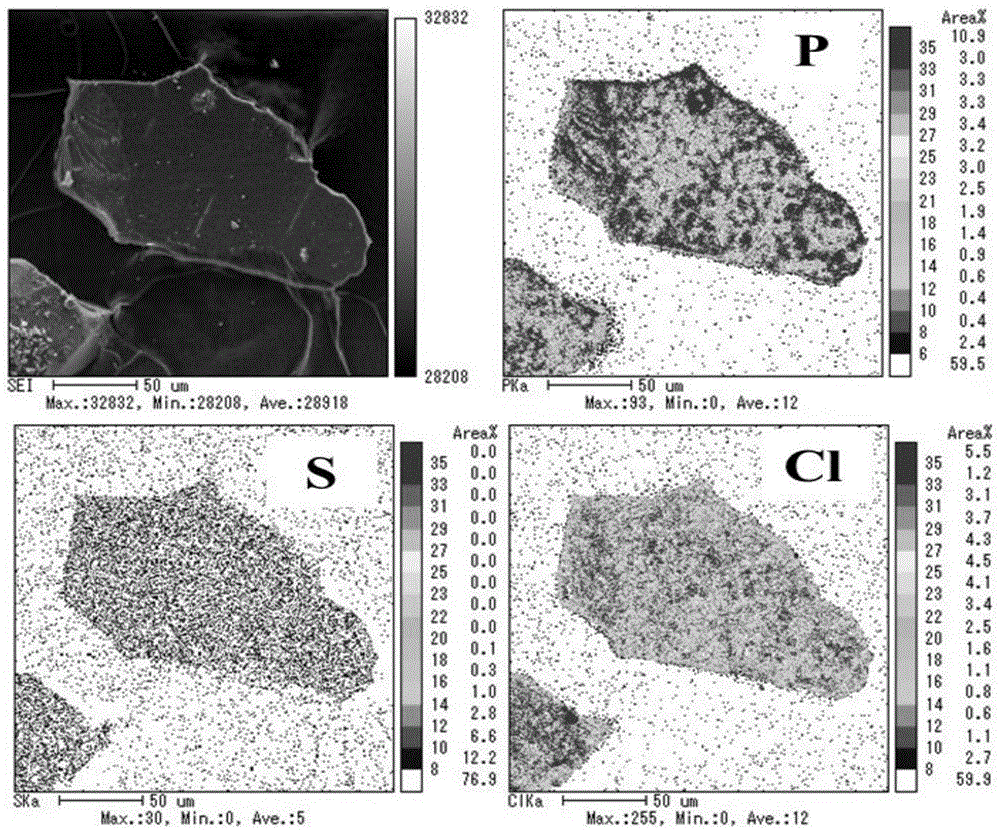
将三元水滑石吸附剂mgmnzr-ldh吸咐海水中磷酸根后,回收吸附剂,通过epma对吸附剂进行电子探针微区分析,元素分布如图10所示,从图10可以看出,p元素的吸咐量较大,而在海水里cl离子的含量较高的,但是由于p与三元水滑石吸附剂中zr的亲和力较强,即使在海水这种竞争离子较多,并且浓度较高的情况下,三元水滑石吸附剂mgmnzr-ldh依然能选择性地从共存离子的竞争体系中吸附p元素。
吸附海水中po43-后的三元水滑石吸附剂mgmnzr-ldh、mgmnzr-ldo130分别放入浓度为0.1m的氢氧化钠水溶液25℃下震荡9h,过滤出来,完成再生。将再生后的三元水滑石吸附剂mgmnzr-ldh、mgmnzr-ldo130再次吸附海水中po43-,三元水滑石吸附剂mgmnzr-ldh的平衡吸咐量达到15.3mg·g-1,mgmnzr-ldo130的平衡吸咐量达到12.2mg·g-1。再进行再生,再吸咐,五次循环后,三元水滑石吸附剂mgmnzr-ldh的平衡吸咐量达到14.2mg·g-1,mgmnzr-ldo130的平衡吸咐量达到11.5mg·g-1。在多次循环再生后使用仍然具有较好的吸附量,说明结构稳定性高。
实施例4:本实施例4制备的三元水滑石吸附剂mgalzr-ldh的制备方法,按以下步骤进行:
一、按mg(no3)2·6h2o、al(no3)3·9h2o、zrcl2o·8h2o摩尔比6:1:1,将15.38gmg(no3)2·6h2o、3.75gal(no3)3·4h2o、3.22gzrcl2o·8h2o加入100ml水中溶解,得到混合盐溶液d;
二、按氢氧化钠与碳酸钠的摩尔比为2:1,将1.2g氢氧化钠与1.59g碳酸钠加入150ml水中溶解,得到混合溶液b;
三、在搅拌条件下,向步骤一得到的混合盐溶液a中滴加混合溶液b,滴加过程中控制整个溶液ph值在8~10之间,得到混合溶液e;
四、将混合溶液e在80℃的条件下搅拌12h,然后离心分离,将固相物用去离子水洗涤干净,再经在60℃条件下干燥48h、研磨,得到三元水滑石吸附剂mgalzr-ldh。
实施例5:本实施例5的三元水滑石吸附剂mgalzr-ldo的制备方法,按以下步骤进行:
将实施例4制备的三元水滑石吸附剂mgalzr-ldh放在温度为130℃的马弗炉中煅烧4h,得到三元水滑石吸附剂,该三元水滑石吸附剂用mgalzr-ldo130表示。
实施例6:本实施例是实施例5的对比试验,本实施例与实施例5不同的是煅烧温度为500℃,其它与实施例5相同,得到作为对比的三元水滑石,用mgalzr-ldo500表示。
实施例4制备的三元水滑石吸附剂mgalzr-ldh的热失重变化tg-dta如图11所示:通过观察图11中100℃和370℃左右的两个吸热峰,低温部分的吸热峰推断是产物失去游离水,说明产物仍然维持层状结构,而高温测的吸热峰则是层间的水分子以及碳酸跟离子分解,层间只留下了羟基引起的。
实施例4制备的三元水滑石吸附剂mgalzr-ldh、实施例5制备的mgalzr-ldo130和实施例6制备的mgalzr-ldo500的xrd谱图如图12所示,从图12可以看出,不同烧成温度产物的xrd谱图虽然都具有明显的层状结构,但是随着烧成温度的升高衍射峰的强度越来越小,这可能是由于随着温度升高层间的的水分子和碳酸根离子消失或转化为半径较小的羟基,有一部分不定型层形成的原因。并且煅烧500℃之后的谱图衍射峰的位置有了很大偏离,说明层间的羟基消失,水滑石的层结构已经损坏。
实施例4制备的三元水滑石吸附剂mgalzr-ldh的扫描电镜照片如图13所示,实施例5制备的mgalzr-ldo130的扫描电镜照片如图14所示,实施例6制备的mgalzr-ldo500的扫描电镜照片如图15所示,从图14~图15比较可以看出随着煅烧温度的升高,由于层状结构的坍塌,表面的凹凸程度变大,并且表明颗粒状增加。
实施例4制备的三元水滑石吸附剂mgalzr-ldh、实施例5制备的mgalzr-ldo130和实施例6制备的mgalzr-ldo500的比表面积对比如图16所示,随着煅烧温度升高,层状结构的坍塌,水滑石的孔径变大比表面积变小。
三元水滑石吸附剂mgalzr-ldh的比表面积为46.9m2·g-1,孔容积是4.45×10-3cm3·g-1,孔径为10.7nm。mgalzr-ldo130的比表面积为1.19m2·g-1,孔容积是2.81cm3·g-1,孔径为9.43nm。由于在吸附过程较大的比表面积中能够容纳更多的吸附质,另外吸附材料的孔径与磷酸跟离子的大小匹配程度也与吸附量有关,一般是越大吸附越容易。而mgalzr-ldo500由于煅烧温度过高,层状结构的坍塌,水滑石的孔径变大,不利于吸咐。
将实施例4制备的三元水滑石吸附剂mgalzr-ldh、实施例5制备的mgalzr-ldo130和实施例6制备的mgalzr-ldo500应用于po43-吸附,具体按以下步骤进行:
准确称取0.1g/lmgalzr-ldh、mgalzr-ldo130或mgalzr-ldo500加入到ph值为8的含po43-的海水中,其中海水中po43-的浓度为5ppm;将其放入恒温震荡箱中,震荡120min,期间取上层清液,并用磷钼蓝分光光度法测上层清液中磷酸根的浓度,计算吸附量。mgalzr-ldh、mgalzr-ldo130或mgalzr-ldo500的吸附量如图17所示。从图17可以看出,mgalzr-ldh、mgalzr-ldo130吸附磷酸根的效果较好,mgalzr-ldh的平衡吸咐量达到13.7mg/g,mgalzr-ldo130平衡吸咐量达到11.3mg/g,100min以内就能达到吸咐平衡,而mgalzr-ldo500因为层状结构的坍塌,吸附量较低,为8.8mg/g。
为了进一步明确吸附过程中的机理,通过拟一阶和拟二阶动力学方程拟合,结果表明三元水滑石及其焙烧物对磷酸根的吸附过程更符合拟二阶动力学模型,如图18所示。证明吸附过程发生在不止发生在吸附剂表面。
将三元水滑石吸附剂mgalzr-ldh吸咐海水中磷酸根后,回收吸附剂,通过epma对吸附剂进行电子探针微区分析,元素分布如图19所示,从图19可以看出,磷元素均匀吸附在吸附剂上。p元素的吸咐量较大,而在海水里cl离子的含量较高的,但是由于p与三元水滑石吸附剂中zr的亲和力较强,即使在海水这种竞争离子较多,并且浓度较高的情况下,三元水滑石吸附剂mgalzr-ldh依然能选择性地从共存离子的竞争体系中吸附p元素。
吸附海水中po43-后的三元水滑石吸附剂mgalzr-ldh、mgalzr-ldo130分别放入浓度为0.1m的氢氧化钠水溶液25℃下震荡9h,过滤出来,完成再生。将再生后的三元水滑石吸附剂mgalzr-ldh、mgalzr-ldo130再次吸附海水中po43-,三元水滑石吸附剂mgalzr-ldh的平衡吸咐量达到12.7mg·g-1,mgalzr-ldo130的平衡吸咐量达到10.9mg·g-1。再进行再生,再吸咐,五次循环后,三元水滑石吸附剂mgalzr-ldh的平衡吸咐量达到12.2mg·g-1,mgalzr-ldo130的平衡吸咐量达到10.1mg·g-1。在多次循环再生后使用仍然具有较好的吸附量,说明结构稳定性高。
实施例7:本实施例的三元水滑石吸附剂mgmnzr-ldo的制备方法,按以下步骤进行:将实施例1制备的三元水滑石吸附剂mgmnzr-ldh放在温度为280℃的马弗炉中煅烧4h,得到三元水滑石吸附剂,该三元水滑石吸附剂用mgmnzr-ldo280表示。
本实施例的mgmnzr-ldo280对海水中的po43-的平衡吸咐量达到12.6mg·g-1,100min以内就能达到吸咐平衡。再生五次后,平衡吸咐量仍达到11.8mg·g-1,具有很好的稳定性。
实施例8:本实施例的三元水滑石吸附剂mgalzr-ldo的制备方法,按以下步骤进行:将实施例4制备的三元水滑石吸附剂mgalzr-ldh放在温度为300℃的马弗炉中煅烧4h,得到三元水滑石吸附剂,该三元水滑石吸附剂用mgalzr-ldo300表示。
本实施例的mgalzr-ldo300对海水中的po43-的平衡吸咐量达到10.6mg/g,100min以内就能达到吸咐平衡,再生五次后,平衡吸咐量仍达到10.2mg·g-1,具有很好的稳定性。
- 还没有人留言评论。精彩留言会获得点赞!