C3N5材料及其制备方法和应用与流程
本发明涉及c3n5材料及其制备方法和应用,属于光催化技术领域。
背景技术:
光催化技术是一种利用光催化剂在光照射下发生催化反应的技术,是一种在能源和环境领域有着重要应用前景的绿色技术。光催化剂是光催化技术的关键,是在光子的激发下能够起到催化作用的化学物质的统称,半导体光催化剂能够在光的照射下激发产生电子和空穴,在半导体表面发生氧化还原反应,从而实现水的分解或有机污染物的分解,实现光能的利用和转化。
作为一种新型的半导体材料,c3n5以其独特的组成与结构以及可见光催化活性等特点成为研究的热点。与现有的较为研究较多的c3n4材料相比,c3n5具有更高的扩散极限电流密度和更低的过电位,使其光催化性能更佳。目前,c3n5的制备方法有一些报道,但是对其具体结构的报道很少,而不同的方法制备得到的c3n5材料,其光催化活性并不相同,这可能是由于方法不同造成其结构的不同所带来的。
c3n5的制备可以采用5-氨基-1h-四唑(5-attz)自组装制备,比如:inyoungkim等在文献《具有三唑和三嗪骨架的有序介孔c3n5及其石墨烯复合物》中的报道(dr.inyoungkim,sunghokim,dr.xiaoyanjin,dr.selvarajanpremkumar,dr.goutamchandra,dr.nam-suklee,prof.gurudasp.mane,prof.seong-juhwang,prof.sivaumapathy,prof.ajayanvinu.orderedmesoporousc3n5withacombinedtriazoleandtriazineframeworkanditsgraphenehybridsfortheoxygenreductionreaction(orr)[j].angewandtechemie,2018,130(52).)
c3n5的制备也可以采用3-氨基-1,2,4-三唑为原料制备,比如专利cn109562940a中,采用3-氨基-1,2,4-三唑为原料制备得到c3n5材料。该方法制备得到的材料,其光催化性能有待进一步的提高。
而湖南师范大学张友玉等研究了在naoh的辅助下,3-氨基-1,2,4-三唑为原料制备得到c3n5材料,发现随着naoh浓度的增加,g-c3n5的n空位逐渐增大,表现出优异的光催化和光电化学性能。(详见文献《具有可控氮空位的多孔石墨氮化碳:增强污染物可见光降解的理想催化剂》,haiyanwang,mingxiali,huanli,qiujunlu,youyuzhang,shouzhuoyao.porousgraphiticcarbonnitridewithcontrollablenitrogenvacancies:aspromisingcatalystforenhanceddegradationofpollutantundervisiblelight[j].materials&;design,2018.)此外,他们还研究了在kbr的辅助下,3-氨基-1,2,4-三唑为原料制备得到c3n5材料,发现与直接制备得到的材料相比,在kbr辅助下得到的c3n5材料也显示出显着优异的光催化性能。(详见文献《盐导向合成的介孔棒状g-c3n5:作为降解有机污染物的优良光催化剂》,wang,haiyan,li,mingxia,lu,qiujun,etal.amesoporousrod-likeg-c3n5synthesizedbysalt-guidedstrategy:asasuperiorphotocatalystfordegradationoforganicpollutant[j].acssustainablechemistry&engineering.)
技术实现要素:
本发明解决的第一个技术问题是提供一种c3n5材料的制备方法,采用该方法制备得到的材料,与现有的方法相比,表现出更优的光催化性能。
本发明c3n5材料的制备方法,包括如下步骤:
a、混料:将3-氨基-1,2,4-三唑和六元氮杂环按重量比1:0.8~1.2混匀,得到混匀后的粉末,其中,六元氮杂环为三聚氰酸和巴比妥酸的至少一种;
b、煅烧:将混匀后的粉末升温,于480~520℃保温2.5~3.5h,冷却后取出,洗涤,干燥,得到c3n5材料。
优选的,a步骤的具体操作为:将3-氨基-1,2,4-三唑和六元氮杂环加入无水乙醇中,搅拌1~3h,然后超声处理0.5~1.5h,60~80℃干燥,得到混匀后的粉末。更优选搅拌2h,然后超声处理1h,70℃干燥。
优选的,a步骤中,六元氮杂环为三聚氰酸。更优选的,3-氨基-1,2,4-三唑和三聚氰酸按重量比1:1混匀。
另一优选的,a步骤中,六元氮杂环为三聚氰酸和巴比妥酸。更优选的,按重量比,3-氨基-1,2,4-三唑:三聚氰酸:巴比妥酸=1:1:0.1。
优选的,b步骤中,升温速率为3~8℃/min,优选升温速率为5℃/min。
作为优选方案,a步骤之后进行c步骤,所述c步骤为:将混匀后的粉末加入助剂溶液中,并搅拌均匀,然后干燥,再将干燥后的粉末进行b步骤;所述助剂为氢氧化钠或溴化钾。
优选的,所述助剂溶液的浓度为0.05~0.15g/ml,更优选所述助剂溶液的浓度为0.1g/ml。
优选的,助剂与3-氨基-1,2,4-三唑的重量比为1:10~20,更优选助剂与3-氨基-1,2,4-三唑的重量比为1:15。
本发明解决的第二个技术问题是提供一种c3n5材料。
本发明c3n5材料,采用上述方法制备得到。通过紫外漫反射和荧光发射光谱可以看出,该材料与现有方法制备得到的c3n5材料相比,其光催化活性更好。
本发明还提供本发明所述的c3n5材料在光催化剂中的应用。
本发明的c3n5材料,其光催化活性较好,可用于光催化剂中。本发明c3n5材料可以单独作为光催化剂使用,也可以与其他光催化剂混合使用。
与现有技术相比,本发明具有如下有益效果:
本发明在制备c3n5材料时,采用3-氨基-1,2,4-三唑和特定的六元氮杂环混合作为原料,其制备方法简单,由此得到的c3n5材料,其光催化活性较好,可广泛用作光催化剂。
附图说明
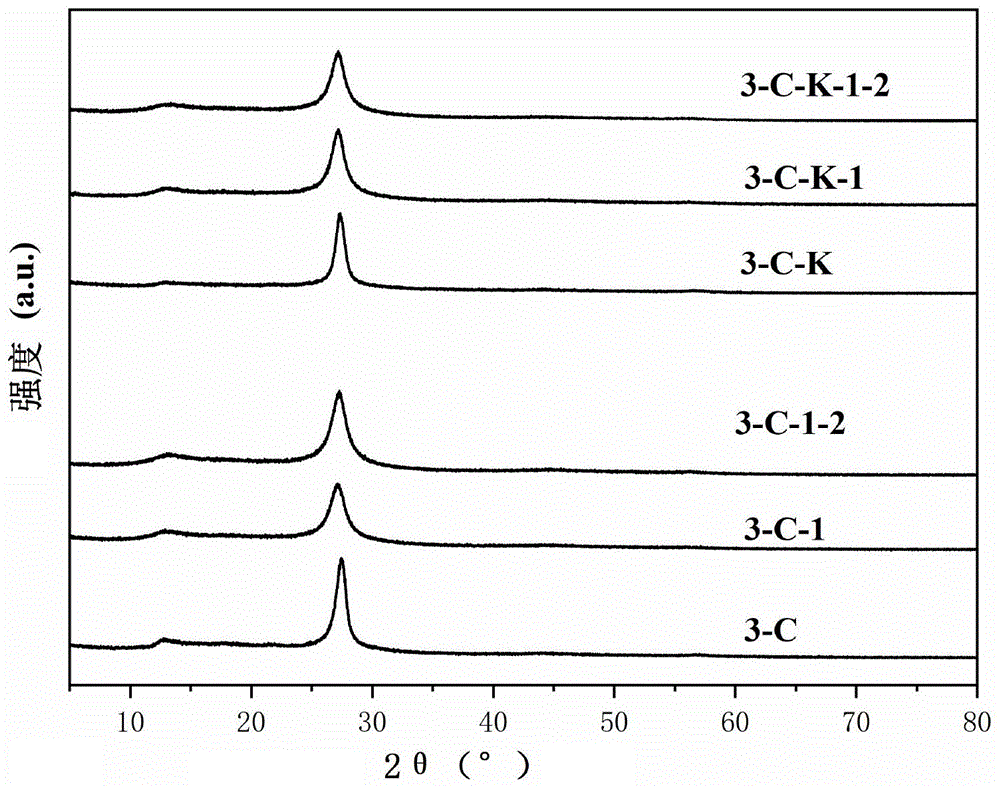
图1为本发明实施例1~4和对比例1~2制备得到的c3n5的xrd图谱。
图2为本发明实施例1~4和对比例1~2制备得到的c3n5的紫外漫反射图谱。
图3为本发明实施例1~4和对比例1~2制备得到的c3n5在366nm激发下的荧光发射图谱。
具体实施方式
本发明c3n5材料的制备方法,包括如下步骤:
a、混料:将3-氨基-1,2,4-三唑和六元氮杂环按重量比1:0.8~1.2混匀,得到混匀后的粉末,其中,六元氮杂环为三聚氰酸和巴比妥酸的至少一种;
b、煅烧:将混匀后的粉末升温于480~520℃保温2.5~3.5h,冷却后取出,洗涤,干燥,得到c3n5材料。
a步骤主要是为了将原料按配比混匀,优选的,a步骤的具体操作为:将3-氨基-1,2,4-三唑和六元氮杂环加入无水乙醇中,搅拌1~3h,然后超声处理0.5~1.5h,60~80℃干燥,得到混匀后的粉末;优选搅拌2h,然后超声处理1h,70℃干燥。
作为其中一种实施方式,a步骤中,六元氮杂环为三聚氰酸。优选的,3-氨基-1,2,4-三唑和三聚氰酸按重量比1:1混匀。
作为另一种实施方式,a步骤中,六元氮杂环为三聚氰酸和巴比妥酸。研究表明,加入3-氨基-1,2,4-三唑、三聚氰酸和巴比妥酸后得到的c3n5材料的光催化活性优于在制备时仅加入3-氨基-1,2,4-三唑和三聚氰酸的,即巴比妥酸的加入,有利于提高合成的材料的光催化性能。优选的,按重量比,3-氨基-1,2,4-三唑:三聚氰酸:巴比妥酸=1:1:0.1。
b步骤为煅烧得到c3n5材料的过程。该步骤的煅烧无需保护气体,可在空气中进行,操作简单,对煅烧设备没有特殊的要求,比如,可以直接将混匀后的粉末放入马弗炉中进行升温煅烧。
优选的,b步骤中,升温速率为3~8℃/min,优选升温速率为5℃/min。
作为优选方案,a步骤之后进行c步骤,所述c步骤为:将混匀后的粉末加入助剂溶液中,并搅拌均匀,然后干燥,再将干燥后的粉末进行b步骤;所述助剂为氢氧化钠或溴化钾。加入助剂后,得到的c3n5材料的性能更佳。
优选的,所述助剂溶液的浓度为0.05~0.15g/ml,更优选所述助剂溶液的浓度为0.1g/ml。
优选的,助剂与3-氨基-1,2,4-三唑的重量比为1:10~20,更优选助剂与3-氨基-1,2,4-三唑的重量比为1:15。
本发明c3n5材料,采用上述方法制备得到。通过紫外漫反射和荧光发射光谱可以看出,该材料与现有方法制备得到的c3n5材料相比,其光催化活性更好。
本发明还提供本发明所述的c3n5材料在光催化剂中的应用。
本发明的c3n5材料,其光催化活性较好,可用于光催化剂中。本发明c3n5材料可以单独作为光催化剂使用,也可以与其他光催化剂混合使用。
下面结合实施例对本发明的具体实施方式做进一步的描述,并不因此将本发明限制在所述的实施例范围之中。
实施例1
分别称取5g的3-氨基-1,2,4-三唑和5g的三聚氰酸装入烧杯并加入100ml无水乙醇,接着将悬浮液在环境温度下搅拌2h,然后在室温下超声处理1h。然后将悬浮液放入70℃的烘箱内干燥后得到粉末,将其装入50ml的带盖坩埚中,移至马弗炉以5℃/min的升温速率升至500℃后保温3h。冷却后将样品装入烧杯中并加入100ml的蒸馏水,然后放入超声机中超声分散3h,随后将样品离心并干燥,得到c3n5材料,记为3-c-1。该材料的xrd图谱见图1,紫外漫反射图谱见图2,366nm激发下的荧光发射图谱见图3。
实施例2
分别称取5g的3-氨基-1,2,4-三唑、5g的三聚氰酸和0.5g的巴比妥酸装入烧杯并加入100ml无水乙醇,接着将悬浮液在环境温度下搅拌2h,然后在室温下超声处理1h。然后将悬浮液放入70℃的烘箱内干燥后得到白色粉末,将其装入50ml的带盖坩埚中,移至马弗炉以5℃/min的升温速率升至500℃后保温3h。冷却后将样品装入烧杯中并加入100ml的蒸馏水,然后放入超声机中超声分散3h,随后将样品离心并干燥,得到c3n5材料,记为3-c-1-2。该材料的xrd图谱见图1,紫外漫反射图谱见图2,366nm激发下的荧光发射图谱见图3。
实施例3
分别称取4.5g的3-氨基-1,2,4-三唑和4.5g的三聚氰酸装入烧杯并加入100ml无水乙醇,接着将悬浮液在环境温度下搅拌2h,然后在室温下超声处理1h。然后将悬浮液放入70℃的烘箱内干燥后得到粉末,加入kbr溶液(0.3g的kbr加入3ml蒸馏水)并搅拌均匀,随后放入80℃的烘箱内烘干得到固体,将其移入50ml的带盖坩埚中,移至马弗炉以5℃/min的升温速率升至500℃后保温3h。冷却后将样品装入烧杯中并加入100ml的蒸馏水,然后放入超声机中超声分散3h,将样品洗涤数次后离心并干燥,得到c3n5材料,记为3-c-k-1。该材料的xrd图谱见图1,紫外漫反射图谱见图2,366nm激发下的荧光发射图谱见图3。
实施例4
分别称取4.5g的3-氨基-1,2,4-三唑、4.5g的三聚氰酸和0.45g的巴比妥酸装入烧杯并加入100ml无水乙醇,接着将悬浮液在环境温度下搅拌2h,然后在室温下超声处理1h。然后将悬浮液放入70℃的烘箱内干燥后得到粉末,加入kbr溶液(0.3g的kbr加入3ml蒸馏水)并搅拌均匀,随后放入80℃的烘箱内烘干得到固体,将其移入50ml的带盖坩埚中,移至马弗炉以5℃/min的升温速率升至500℃后保温3h。冷却后将样品装入烧杯中并加入100ml的蒸馏水,然后放入超声机中超声分散3h,将样品洗涤数次后离心并干燥,得到c3n5材料,记为3-c-k-1-2。该材料的xrd图谱见图1,紫外漫反射图谱见图2,366nm激发下的荧光发射图谱见图3。
对比例1
称取6g的3-氨基-1,2,4-三唑装入50ml的带盖坩埚中,移至马弗炉以5℃/min的升温速率升至500℃后保温3h。冷却后将样品装入烧杯中并加入100ml的蒸馏水,然后放入超声机中超声分散3h,随后将样品离心并干燥,得到c3n5材料,记为3-c。该材料的xrd图谱见图1,紫外漫反射图谱见图2,366nm激发下的荧光发射图谱见图3。
对比例2
称取4.5g的3-氨基-1,2,4-三唑装入烧杯中,加入kbr溶液(0.3g的kbr加入3ml蒸馏水)并搅拌均匀,随后放入80℃的烘箱内烘干得到固体,将其移入50ml的带盖坩埚中,移至马弗炉以5℃/min的升温速率升至500℃后保温3h。冷却后将样品装入烧杯中并加入100ml的蒸馏水,然后放入超声机中超声分散3h,将样品洗涤数次后离心并干燥,得到c3n5材料,记为3-c-k。该材料的xrd图谱见图1,紫外漫反射图谱见图2,366nm激发下的荧光发射图谱见图3。
图1为本发明实施例1~4和对比例1~2制备得到的c3n5的xrd图谱,从图1中可以看出,与现有的方法制备得到的c3n5材料(即对比例1~3)相比,本发明方法制备得到的材料测试xrd时衍射峰与现有方式制备的c3n5材料衍射峰位置一致,可见,本发明成功制备得到了c3n5材料,且本发明所得c3n5材料的xrd衍射峰比现有方法制备得到的c3n5材料衍射峰更宽,说明晶型有所下降,比表面积有所提高,利于光催化性能的提高。
图2为本发明实施例1~4和对比例1~2制备得到的c3n5的紫外漫反射图谱,从图谱上可以看出,与现有方法制备得到的c3n5材料相比,本发明方法制备得到的材料吸收光谱范围更宽,更有利于在可见光或日光下实现光催化。
图3为本发明实施例1~4和对比例1~2制备得到的c3n5在366nm激发下的荧光发射图谱。本领域技术人员公知,光催化剂受光激发后会产生电子和空穴,其中一部分电子和空穴起氧化还原作用,另一部分则会复合,能量以光的形式释放出来,而一般的,电子和空穴复合时会发出荧光,因此,荧光发射强度越低,电子空穴复合率则越低,表明光催化活性越高。而从图3可以看出,本发明方法制备得到的c3n5材料,荧光发射强度均低于现有方法制备得到的c3n5材料,表明本发明方法得到的c3n5材料的光催化活性较高。3-c-1-2的光催化活性优于3-c-1,3-c-k-1-2的光催化活性优于3-c-k-1,表明在制备时加入了巴比妥酸和三聚氰酸的c3n5材料光催化活性高于仅加入了三聚氰酸的。此外,3-c-k-1的光催化活性优于3-c-1,3-c-k-1-2的光催化活性优于3-c-1-2,表明助剂溴化钾的加入有助于提高材料的光催化活性。
- 还没有人留言评论。精彩留言会获得点赞!