Zr(Ⅳ)-2-氨基对苯二甲酸配合物杂化聚合物整体柱的制备方法及其在固相微萃取应用与流程
本发明涉及新型金属-有机配合物杂化聚合物整体柱制备领域,尤其涉及一种zr(ⅳ)-2-氨基对苯二甲酸配合物杂化聚合物整体柱的制备方法及其在固相微萃取应用。
背景技术:
近年来,多孔材料在储能、催化和分离等领域受到越来越多的关注。杂化整体柱是在硅胶整体柱的基础上发展起来的,在有机溶剂中稳定,且具有较好的机械稳定性,能够减少材料溶胀和断裂现象。因此,有机-无机杂化整体柱的制备与应用成为了现代分离学科的又一研究热点,但是目前有机-无机杂化整体柱对物质的吸附量低,不能满足对痕量物质的微萃取的需要。
金属-有机配合物中存在金属的空轨道和配体的杂原子孤对电子,能够和许多有机小分子通过分子间作用进行结合,而且该类材料比表面积大,所以,金属-有机配合物有望成为新型吸附剂的表面修饰材料。
技术实现要素:
为了解决以上问题,并开发金属-有机配合物的新应用,本发明的目的是提供一种zr(ⅳ)-2-氨基对苯二甲酸配合物杂化聚合物整体柱(oipm-zratp),增大了材料的比表面积和吸附能力,提高了材料的吸附容量。
为实现上述目的,本发明所设计的zr(ⅳ)-2-氨基对苯二甲酸配合物杂化聚合物整体柱的制备方法,其特征在于,包括步骤:首先制备有机-无机杂化聚合物整体柱,然后将zrcl4溶液流经有机-无机杂化聚合物整体柱,然后再将2-氨基对苯二甲酸溶液流经有机-无机杂化聚合物整体柱,最后用甲醇冲洗有机-无机杂化聚合物整体柱,即完成一个循环组装过程;进行一次或一次以上的循环自组装反应,即得zr(ⅳ)-2-氨基对苯二甲酸配合物杂化聚合物整体柱。
与现有的有机-无机杂化聚合物整体柱相比,本发明通过层层自组装法,在有机-无机杂化聚合物整体柱(oipm)的表面均匀连续生长zr(ⅳ)-2-氨基对苯二甲酸配合物(zratp),增大了oipm的比表面积,提高了材料的吸附容量,同时,改善了整体柱的吸附性能。
作为优选方案,所述zrcl4溶液的浓度为5~15mmoll-1,所述zrcl4溶液的流速为0.01~0.03mlmin-1,单次循环自组装过程体积为1.0~2.0ml;所述2-氨基对苯二甲酸溶液的浓度为5~15mmoll-1,所述2-氨基对苯二甲酸溶液的流速为0.01~0.03mlmin-1,单次循环自组装过程体积为1.0~2.0ml。
作为优选方案,所述有机-无机杂化聚合物整体柱的制备过程为,首先,将甲基三甲氧基硅烷、甲醇、硝酸混合反应后再加入3-(甲基丙烯酰氧基)丙基三甲氧基硅烷和甲醇的混合溶液,搅拌均匀得到无机醇解液;然后将2,2-偶氮二异丁腈、甲醇、甲基丙烯酸、乙二醇二甲基丙烯酸酯混合得到有机预聚液;最后将无机醇解液和有机预聚液混合,在密封的反应容器内反应,洗涤后得到有机-无机杂化聚合物整体柱。
作为优选方案,所述无机醇解液的制备具体过程为,将甲基三甲氧基硅烷300~500μl、100~200μl甲醇、100~200μl0.1moll-1硝酸,混合反应后再加入100~200μl3-(甲基丙烯酰氧基)丙基三甲氧基硅烷和甲醇的混合溶液,混合溶液中3-(甲基丙烯酰氧基)丙基三甲氧基硅烷和甲醇的体积比为1:1~2;所述有机预聚液的制备具体过程为,将10~15mg2,2-偶氮二异丁腈、1~2ml甲醇、30~35μl0.2mmol甲基丙烯酸、300~400μl2mmol乙二醇二甲基丙烯酸酯混合;所述无机醇解液和有机预聚液的体积比为1:2~4。
一种zr(ⅳ)-2-氨基对苯二甲酸配合物杂化聚合物整体柱的应用,其特征在于,所述zr(ⅳ)-2-氨基对苯二甲酸配合物杂化聚合物整体柱对带有磷酸基团目标物固相微萃取中的应用。
作为优选方案,所述zr(ⅳ)-2-氨基对苯二甲酸配合物杂化聚合物整体柱应用于猪肝样品中磷酸化丝氨酸的微萃取。利用oipm-zratp能够提高猪肝样品中磷酸化丝氨酸检测灵敏度。
作为优选方案,所述zr(ⅳ)-2-氨基对苯二甲酸配合物杂化聚合物整体柱微萃取磷酸化丝氨酸的过程为,在微量注射泵的推动下,将带有磷酸化丝氨酸的猪肝样品从注射器内流过zr(ⅳ)-2-氨基对苯二甲酸配合物杂化聚合物整体柱,磷酸化丝氨酸被吸附在zr(ⅳ)-2-氨基对苯二甲酸配合物杂化聚合物整体柱上,然后利用洗脱剂将磷酸化丝氨酸洗脱下来得到洗脱液,最后利用高效液相色谱-串联质谱技术检测分析洗脱液中磷酸化丝氨酸的含量。
附图说明
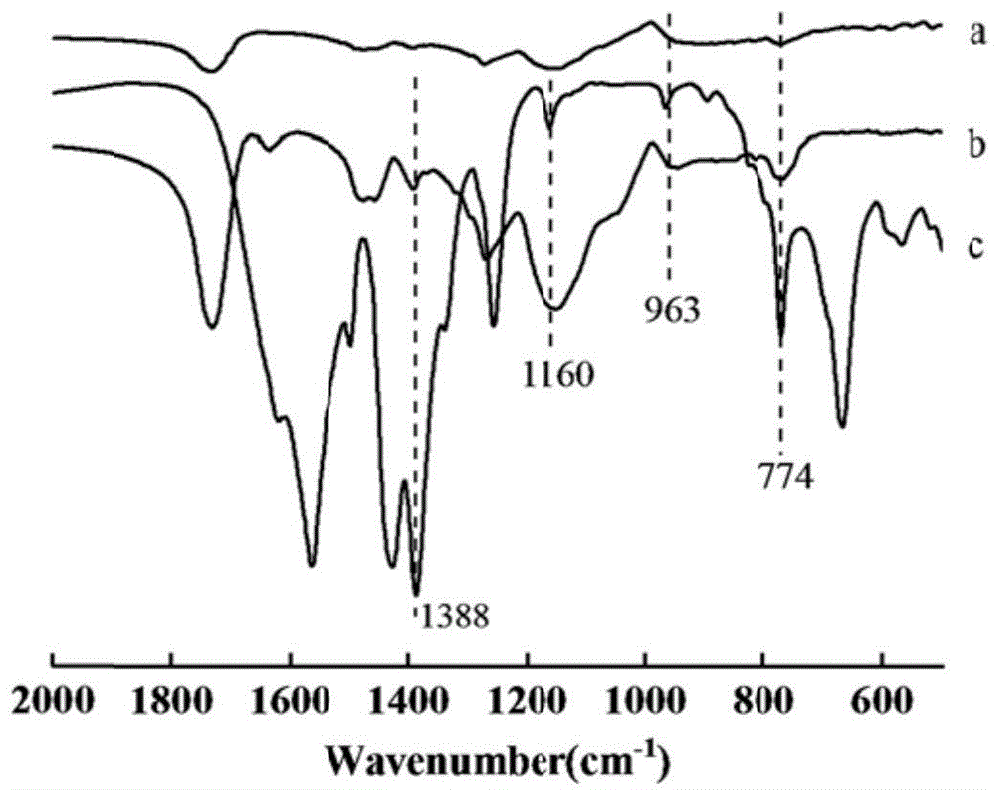
图1为opim-zratp和未组装的opim以及uio-66-nh2的红外图谱图;
图2a为opim的氮吸附曲线图;
图2b为opim-zratp的氮吸附曲线图;
图3a为opim的孔径分布图;
图3b为opim-zratp的孔径分布图;
图4a为oipm放大1000倍的sem图;
图4b为oipm放大3000倍的sem图;
图4c为opim-zratp放大1000倍的sem图;
图4d为opim-zratp放大3000倍的sem图;
图5为oipm-zratp对磷酸化丝氨酸(p-ser)的吸附曲线图;
图6为猪肝样品中磷酸化丝氨酸(p-ser)的色谱图。
具体实施方式
为更好地理解本发明,以下将结合附图和具体实例对发明进行详细的说明。
为解决现有杂化整体柱对目标物质吸附量低的问题,本发明提供一种zr(ⅳ)-2-氨基对苯二甲酸配合物杂化聚合物整体柱的制备方法,其以有机-无机杂化聚合物整体柱(oipm)为载体,通过层层自组装法,使zr(ⅳ)-2-氨基对苯二甲酸配合物(zratp)在载体上均匀连续生长,增加了oipm的比表面积,提高了材料的吸附容量。以下将通过具体的实施例来对本发明的zr(ⅳ)-2-氨基对苯二甲酸配合物杂化聚合物整体柱的制备方法的优选方式进行详细地说明。
实施例
zr(ⅳ)-2-氨基对苯二甲酸配合物杂化聚合物整体柱的制备方法,包括步骤:
(1)无机醇解液的制备:在离心管中依次加入400μl甲基三甲氧基硅烷(mtms),150μl甲醇和100μl0.1moll-1硝酸,旋涡2min,40℃下反应1h,再加入150μl3-(甲基丙烯酰氧基)丙基三甲氧基硅烷(γ-maps)和甲醇(1:1)的混合溶液,搅拌反应1h。
(2)有机预聚液的制备:向另一离心管中加入12mg2,2-偶氮二异丁腈(aibn),1ml甲醇,0.2mmol甲基丙烯酸(34μl)和2mmol乙二醇二甲基丙烯酸酯(egdma)(377μl),旋涡溶解aibn,并混匀溶液。
(3)有机-无机杂化聚合物整体柱的制备:取体积比1:3的无机醇解液和有机预聚液混匀,超声5min后,向一端封口的移液枪枪头中加入该混合溶液100μl,枪头另一端用硅胶密封,继续超声5min,除去底端气泡,最后将移液枪枪头置于60℃的恒温箱中反应24h,得到有机-无机杂化聚合物整体柱(opim),待反应完成后除去硅胶,用剪刀减去封口端,用甲醇冲洗除去残余物,泡在甲醇中备用。
(4)有机-无机杂化聚合物整体柱的修饰:先用微量注射泵将1.2ml10mmoll-1zrcl4溶液推至有机-无机杂化聚合物整体柱,流速为0.02mlmin-1,时间为1h,再用1.2ml10mmoll-12-氨基对苯二甲酸(atp)推至有机-无机杂化聚合物整体柱,流速为0.02mlmin-1,最后以0.02mlmin-1的流速甲醇冲洗,即完成一个循环自组装过程,进行十个循环自组装过程后得到zr(ⅳ)-2-氨基对苯二甲酸配合物杂化聚合物整体柱(opim-zratp),用甲醇冲洗残余物,将opim-zratp泡在甲醇中备用。
将本实施例得到的opim-zratp与opim以及uio-66-nh2进行红外图谱分析,结果如图1所示,其中a曲线表示opim的红外图谱,b曲线表示opim-zratp的红外图谱,c曲线表示uio-66-nh2的红外图谱,将b曲线与a曲线比较,b曲线中出现了zr-o的1160cm-1和963cm-1处伸缩振动峰,表明zr(ⅳ)成功与羧基配位,将b曲线与c曲线比较,1388cm-1、774cm-1峰分别对应于羧基中c-o的伸缩振动和-nh2的摇摆弯曲振动,且与c中的吸收峰相对应,表明聚合物中atp配位成功。因此,从图1中可以看出opim-zratp合成成功。
将本实施例得到的opim-zratp与opim分别进行孔径分布测试和氮吸附测试,结果如图2a、图2b和图3a和图3b。从图2a、图2b可以看出,本实施例的opim-zratp比表面积为93.8m2g-1,大于oipm材料的比表面积(11.8m2g-1),从图3a和图3b可以看出,oipm-zratp材料的孔径主要分布在4nm左右,说明oipm-zratp材料为介孔材料。进一步结合扫描电子显微镜表征opim-zratp物理形貌,结合图4a、图4b、图4c、图4d可以看出,opim-zratp材料的表面比oipm材料表面粗糙,而且从图4c和图4d可以看出,opim-zratp材料表面分布了较多聚合物小颗粒,即经过层层自组装后,zratp均匀覆盖在oipm表面,有效增加材料的比表面积。
实施例2
利用zr(ⅳ)-2-氨基对苯二甲酸配合物杂化聚合物整体柱微萃取生物样品中磷酸化丝氨酸(p-ser)的过程为,将实施例1得到的opim-zratp直接牢固卡扣在注射器上(实施例1得到的opim-zratp固定在移液枪枪头中,将移液枪枪头卡扣在注射器上),将样品溶液置于注射器内,在微量注射泵的推动下,样品从注射器内流过opim-zratp,p-ser被吸附在opim-zratp上,然后用醋酸-醋酸钠缓冲液洗脱opim-zratp上p-ser得到洗脱液,然后利用利用高效液相色谱-串联质谱技术检测洗脱液中p-ser的含量,进而得到opim-zratp对p-ser的吸附量。
分别配制p-ser浓度为50μgml-1、100μgml-1、150μgml-1、200μgml-1、300μgml-1的样品溶液,经opim-zratp吸附、脱附后利用利用高效液相色谱-串联质谱技术检测洗脱液中p-ser的含量进而得到opim-zratp对p-ser的吸附量。结合图5所示,opim-zratp对p-ser的饱和吸附量约为20mgg-1,说明材料对p-ser吸附性能较好。
利用实施例1得到的opim-zratp检测猪肝样品中p-ser,该方法的线性范围、检出限和定量限见表,可以看出,本方法线性范围宽,对猪肝样品中p-ser的检出限lod(s/n=3)为2.2ngg-1,本方法能够应用于肝脏样品p-ser的灵敏检测,与文献“steinfeldjb,aernihr,rogulinas,etal.expandedcellularaminoacidpoolscontainingphosphoserine,phosphothreonine,andphosphotyrosine[j].acschemicalbiology,2014,9(5):1104-1112”报道的用于检测p-ser的hplc-ms/ms方法比较,本申请检出限2.2ngg-1远低于文献中的检测限6.2μgg-1,说明本实验的中opim-zratp富集效果好,能有效提高p-ser的检测灵敏度。
表1线性范围、检出限和定量限
在线性范围内,分别以高、中、低三个加标磷酸化丝氨酸p-ser浓度的样品进行spme-hplc-ms/ms分析,考察方法的精密度和回收率,结果见表。可见,日内rsd在2.8-5.7%范围内,日间rsd在2.9-5.1%范围内,加标回收率为93.6-104.0%。因此,本申请重现性好、回收率和灵敏度高、可用于猪肝样品中p-ser含量的分析。
表2精密度和回收率
分别制备空白猪肝样品(a)、向猪肝样品中添加500ngg-1p-ser(b)、猪肝样品经opim-zratp萃取(c),进行lc-ms/ms检测,结果如图6所示,从图6可以看出,利用opim-zratp对猪肝样品中的p-ser有很好的富集效果。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
- 还没有人留言评论。精彩留言会获得点赞!