制备光气的催化剂及其制备方法和光气制备与能源综合利用的方法与流程
本发明属于光气合成领域,更具体的涉及一种制备光气的催化剂及光气合成能源综合利用的技术。
背景技术:
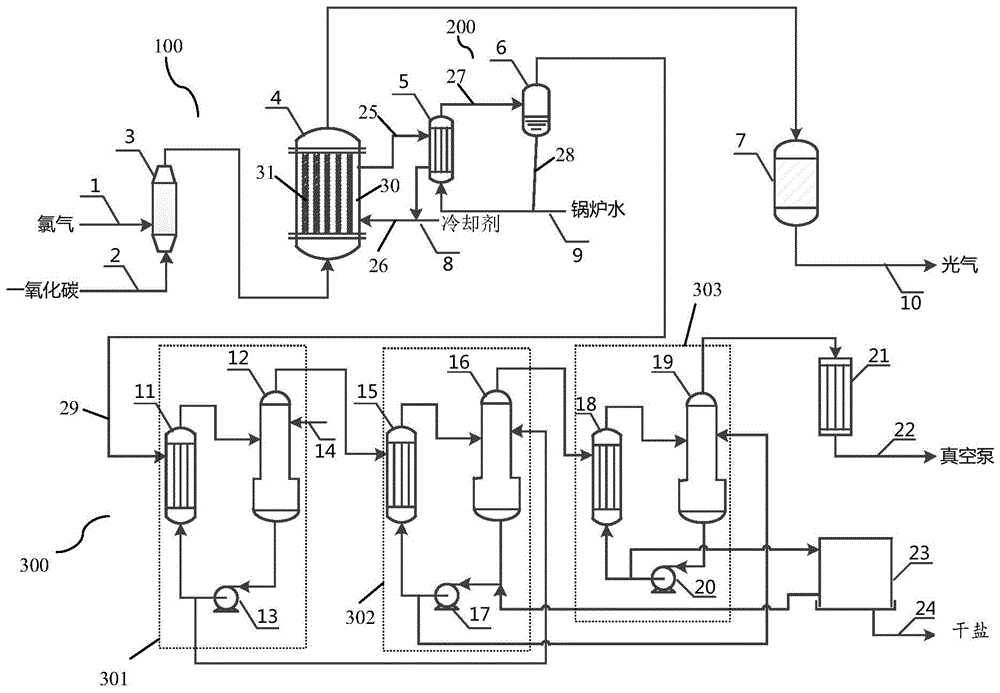
:光气学名碳酰氯,烂苹果味,纯光气无色,工业产品显浅黄色或浅绿色,由于具有两个酰氯,化学性质十分活泼。光气是一种非常重要的化工原料,在有机合成上有着广泛的用途,它广泛应用于农药、染料、涂料、引发剂、医药、精细化学品、异氰酸酯(r-nco)、二苯基甲烷二异氰酸酯(mdi)、甲苯二异氰酸酯(tdi)、聚氨酯(pu)等的合成。随着近几年mdi、tdi、pu及专用精细光气化学品市场的蓬勃发展,作为主要原料的光气,其需求也迅速增长。因此,对光气的产量、质量、及其能源综合利用的研究显得尤为重要。工业化制造光气的方法主要是用一氧化碳和氯气作原料,以活性炭为催化剂合成光气,常用的活性炭是椰壳炭和煤基炭。光气合成反应属强放热反应,单位氯气反应热为116kj/mol。对于此类放热反应,反应热是否及时移走是非常关键的。如果反应热不能被及时移走,不仅会造成“飞温”,其绝热温升可达550℃以上,使反应速度加剧,活性炭烧蚀粉化严重;若造成恶性循环,甚至使反应失控。就光气合成反应而言,高温会使光气重新分解为一氧化碳和氯气,光气分解率可达80%以上,过量的氯气会导致光气合成反应器出口的游离氯超标,严重影响光气产品质量。在光气化反应环境中,过量的氯气会与胺反应生成含氯的化合物。这些含氯的杂质化合物很难用常规方法如精溜、重结晶等去除,严重影响产品质量。另外,反应器和壳体温差增大,由于热膨胀程度不同,从而使设备损坏,如采用钢设备,还会因高温引起氯与铁反应而损坏设备。目前,光气合成反应器主要为管程装有活性炭催化剂、壳程走冷却介质的立式列管式固定床反应器,冷却介质主要为低沸点的水、有机溶剂或高沸点的热油。氯气和一氧化碳提前在混合器混合,从底部进入反应器,冷却介质采用并流或逆流的形式均匀带走热量。业内人士进行过许多尝试以改进光气合成的方法,以提高其经济性和光气质量。ep1135329b1公开了一种低四氯化碳的光气的生产方法,通过使用具有低甲烷含量的一氧化碳原料,减少甲烷和氯气反应生成四氯化碳的量,降低四氯化碳对后续产品质量的影响。实际上,原料一氧化碳中的甲烷含量极低,四氯化碳的生成量很大一部分是由于氯气和活性炭在高温条件下反应生成。us4231959公开了一种光气制备的方法,通过提高一氧化碳的过量率来降低光气中的余氯含量,提高产品质量。文中同时提到反应器壳侧除了用水作冷却介质外,用沸水也是可以的,此时产生可用的蒸汽。但所述蒸汽品质较低,经济性不高,且换热介质为水,存在逐步高温导致设备腐蚀,水泄露到反应空间内与光气反应,进一步加剧腐蚀,安全风险较高。cn109289714a公开了一种光气合成反应催化剂的装填方法,通过在活性炭床层上下两处装填100mm-300mm的较大颗粒瓷球,并覆盖1-2层筛网,降低活性炭的流失。但这种传统的装填方式并不能降低反应速率和放热速率,避免飞温产生,活性炭粉化烧蚀情况仍很严重。cn1765740a公开了一种光气制作方法与设备,氯气和一氧化碳在管壳式反应器中反应,该反应器有许多根反应管和环绕反应器的冷却剂空间,冷却剂蒸发用于发蒸汽。但文中仅对发蒸汽的反应器设备进行介绍,对光气合成反应器的热点温度未进行改善说明,且发蒸汽反应器出口温度更高,活性炭催化剂使用寿命较短。目前业内人士主要对光气合成的经济型和产品质量进行了较多研究改善,对提高光气合成反应器使用周期,降低运行成本以及能源综合利用方面报道较少。就光气合成本身而言,当前工艺存在的一个突出问题为活性炭耐高温、抗氧化性能差,反应器径向温差较大,催化剂中心热点温度无法快速移除,高温下活性炭烧蚀粉化,并与原料氯气以及原料气中的微量氧气反应生成四氯化碳或二氧化碳,造成活性炭损失严重;且原料气中含有100-300mg/nm3氧气,进一步加快活性炭烧蚀粉化,反应器使用周期缩短,装置运行稳定性差。另外,光气中的四氯化碳无法有效分离,导致下游产品质量变差。二苯基甲烷二异氰酸酯/多苯基甲烷多异氰酸酯生产过程中会产生大量废盐水,行业内大多采用废盐水脱除有机物后直接排海,造成盐资源的浪费。技术实现要素:有鉴于此,本发明提供一种用于制备光气的催化剂的制备方法,基于该制备方法,能提高活性炭的耐高温和抗氧化性能,提高催化剂的使用寿命,进而提高装置安全稳定运行水平。本发明还提供光气制备与能源综合利用的方法,在光气制备工艺中,使用本发明的催化剂,具有更长的运行周期,从而在光气生产过程中可避免反复停车更换催化剂,进而能产生稳定的蒸汽;将光气生产与盐水蒸发浓缩工艺相耦合,利用光气生产的反应热为热源获得蒸汽,并将该蒸汽作为热源用于盐水蒸发浓缩,不仅解决了能源浪费,而且避免了废盐水排放造成的资源浪费问题,实现能源综合利用一体化的目的。本发明为达到其目的,采用如下技术方案:本发明一方面提供一种用于制备光气的催化剂的制备方法,所述制备方法包括如下步骤:1)将活性炭在改性溶液中搅拌(例如匀速搅拌)浸泡,之后,向所述改性溶液中加入(例如缓慢加入)二甲基二氯化锡和氧化铬粉末进行反应,再加入经热处理的氧化镍细粉,并进行超声震荡,制得预改性活性炭;所述改性溶液为包括磷酸和硅酸的混合酸溶液;2)将步骤1)得到的预改性活性炭烘干;3)在惰性气体(例如氮气、氩气等本领域常见的惰性气体)氛围保护下,将步骤2)烘干的预改性活性炭进行加热焙烧(例如在高温炉中进行),制得所述催化剂。采用本发明的制备方法制备改性活性炭催化剂,通过向磷酸和硅酸的混合酸溶液中加入二甲基二氯化锡和氧化铬粉末充分反应制得磷酸铬锡和硅酸铬锡,氧化镍经热处理后活性较高,与溶液中磷酸铬锡和硅酸铬锡发生反应,形成-o-ni-o-链桥,通过该链桥横向和纵向键合,不断延伸,形成空间网状结构,最终在活性炭表面形成致密的聚合薄层。因而,采用本发明的制备方法得到的改性活性炭催化剂,在活性炭表面覆盖有磷酸铬锡和硅酸铬锡在-o-ni-o-链桥的横向和纵向键合作用下形成的聚合薄层,大大增强了活性炭的耐高温和抗氧化性能,得到的催化剂具有机械强度高、不易粉化、耐高温、抗氧化性能强的特点。一些实施方式中,步骤1)中,所述改性溶液通过将磷酸搅拌溶解于水(例如为纯水)中得到磷酸溶液,之后向磷酸溶液中加入(例如缓慢加入)硅酸,搅拌均匀后得到所述改性溶液。通过先配制磷酸溶液,后加入硅酸来制备改性溶液,能带来更好的混合效果。一些实施方式中,所述磷酸溶液的质量浓度为5%-20%(例如5%、7%、10%、13%、15%、18%、20%),优选7%-18%,更优选10%-15%。一些实施方式中,所述硅酸的用量和所述磷酸的用量的质量比为1:1-1:5(例如1:1、1:2、1:2.5、1:3、1:3.5、1:4、1:5),优选1:2-1:3.5。一些实施方式中,步骤1)中,所述活性炭选自木质活性炭和/或椰壳活性炭,粒径为2mm-7mm(例如2mm、3mm、4mm、5mm、7mm),优选为3mm-5mm。一些实施方式中,步骤1)中,活性炭与改性溶液的质量比为1:0.6-1:2(例如1:0.6、1:0.8、1:1、1:1.2、1:1.5、1:1.8、1:2),优选为1:0.8-1:1.5;活性炭在改性溶液中的浸泡时间为5h-20h(例如5h、10h、13h、15h、18h、20h),优选为10h-15h;一些实施方式中,步骤1)中,所述搅拌的速率为20r/min-50r/min,优选为30r/min-40r/min。一些实施方式中,步骤1)中所述二甲基二氯化锡的加入量为0.2-2.0mol/l(例如0.2mol/l、0.3mol/l、0.5mol/l、0.7mol/l、1.0mol/l、1.3mol/l、1.5mol/l、2.0mol/l),优选为0.3-1.5mol/l,更优选为0.5-1.0mol/l;步骤1)中所述氧化铬的加入量为0.2-3.0mol/l(0.2mol/l、0.3mol/l、0.5mol/l、0.8mol/l、1.0mol/l、1.3mol/l、1.5mol/l、2.0mol/l、2.5mol/l、3.0mol/l),优选为0.5-2.0mol/l,更优选为0.8-1.5mol/l;步骤1)中所述的加入二甲基二氯化锡和氧化铬粉末进行反应的反应时间为1-5h(1h、2h、3h、4h、5h),优选为2-3h。一些实施方式中,步骤1)中,所述氧化镍细粉在温度为400-600℃(例如400℃、450℃、500℃、550℃、600℃),优选为450-550℃下进行所述热处理,能改善活性炭表面负载物的附着和活性炭的机械强度;热处理时间为1-4h(例如1h、2h、3h、4h),优选2-3h。一些实施方式中,步骤1)中,所述氧化镍细粉的粒径为0.3-1.5μm(例如0.3μm、0.5μm、0.6μm、0.8μm、1.0μm、1.2μm、1.5μm),优选为0.5-0.8μm;所述氧化镍细粉的加入量为0.2-1.5mol/l(0.2mol/l、1.5mol/l、0.4mol/l、0.6mol/l、0.8mol/l、1.0mol/l、1.2mol/l、1.5mol/l),优选为0.6-1.2mol/l;步骤1)中加入氧化镍细粉进行反应的反应时间为1-5h(1h、2h、3h、4h、5h),优选为2-3h;所述超声震荡时间与加入氧化镍细粉的反应时间一致。一些实施方式中,步骤1)中,所述超声震荡的超声波脉冲频率为10-30khz(例如10khz、12khz、15khz、17khz、20khz、30khz),优选12-25khz,更优选为15-20khz;脉冲宽度为50-500ms(例如50ms、70ms、100ms、150ms、200ms、250ms、300ms、350ms、400ms、450ms、500ms),优选100-450ms,更优选150-300ms;采用优选条件下的超声震荡,能更好的固化化学改性。如前所述,所述超声震荡时间与加入氧化镍细粉的反应时间一致,优选为1-5h(1h、2h、3h、4h、5h)。一些实施方式中,步骤2)中,进行烘干的温度为150-200℃(例如150℃、170℃、200℃),烘干时间为4h-8h(例如4h、6h、7h、8h)。一些实施方式中,步骤3)中,所述加热焙烧的加热温度为500℃-800℃(例如500℃、550℃、600℃、650℃、700℃、800℃),优选为600℃-700℃;加热时间为5h-15h(例如5h、8h、10h、12h、15h),优选为8h-12h。所述步骤1)中涉及的各操作(浸泡、反应、超声震荡等各环节)均无需对体系温度进行干预控制(例如无需额外的加热等干预),只需在环境温度(或称为室温)下操作即可,例如10-40℃的环境温度下进行所述步骤1)(具体的,例如在后文的实施例1-4中,涉及到步骤1)的各环节,均在25-30℃的环境温度下进行,在后文的实施例1-4中对此将不再赘述)。采用本发明的制备方法制得的催化剂表面覆盖有磷酸铬锡和硅酸铬锡与ni在-o-ni-o-链桥横向和纵向键合作用下形成的聚合薄层,结合超声波的空化作用,促进固化了活性炭的化学改性,大大增强了活性炭的耐高温和抗氧化性能,得到的催化剂具有机械强度高、不易粉化、耐高温、抗氧化性能强的特点。本发明一方面还提供一种用于制备光气的催化剂,所述催化剂以活性炭为载体,所述活性炭表面形成有磷酸铬锡和硅酸铬锡分别与ni通过-o-ni-o-键合而形成的薄层;具体的,载体活性炭表面覆盖有磷酸铬锡和硅酸铬锡在-o-ni-o-链桥横向和纵向键合作用下形成的聚合薄层。该催化剂可采用上文所述的制备方法制得。本发明一方面还提供一种应用,上文所述的催化剂用于催化一氧化碳和氯气进行反应以制备光气;优选的,原料氯气和原料一氧化碳中总的氧含量控制为10-50mg/nm3(例如10mg/nm3、15mg/nm3、20mg/nm3、25mg/nm3、40mg/nm3、50mg/nm3),优选10-20mg/nm3。因原料氯气和一氧化碳中的微量氧气,在高温下会与活性炭反应生成二氧化碳,造成活性炭高温热损失,反应器床层高度下降,使用寿命缩短,反应放热不稳定,影响蒸汽的品质和装置的运行稳定性;本发明人发现,优选将原料氯气和一氧化碳中总的氧含量控制为10-50mg/nm3,更优选为10-20mg/nm3,能有效改善上述弊端,利于延长催化剂寿命,获得稳定的反应放热,从而提高蒸汽品质和装置运行的稳定性。在制备光气时,通过使用本发明制备方法得到的催化剂,其为表面覆盖有磷酸铬锡和硅酸铬锡与ni通过-o-ni-o-链桥横向和纵向键合作用而形成的聚合薄层并经超声波改性固化的活性炭催化剂,具有改善的耐高温和抗氧化性能。在优选方案中,通过使用氧含量为10-20mg/nm3的氯气和一氧化碳来反应制备光气,能极大地降低催化剂与原料氯气及原料气中微量氧气的反应速率,能明显减缓或消除活性炭高温烧蚀或粉化情况,提高催化剂的使用寿命。本发明另一方面还提供一种光气制备与能源综合利用的方法,包括如下步骤:氯气和一氧化碳经混合,进入装填有上文所述的制备方法制得的催化剂或上文所述的催化剂的光气合成反应器内,在所述催化剂的作用下发生反应以合成光气;光气合成反应器内的反应压力可以为0.1-0.5mpag,优选为0.2-0.4mpag;光气合成反应器内的入口温度(co和氯气混合后进反应器的温度)可控制为10-60℃,优选为20-40℃;原料氯气和原料一氧化碳中总氧含量优选控制为10-50mg/nm3(例如10mg/nm3、15mg/nm3、20mg/nm3、25mg/nm3、40mg/nm3、50mg/nm3),更优选10-20mg/nm3;所述光气合成反应器设有冷却剂流通空间(例如光气合成反应器的壳侧,具体为光气合成反应器内围绕列管的空间),所述冷却剂流通空间内流通冷却剂,用于吸收合成光气所产生的反应热;所述冷却剂流通空间内的冷却剂吸收所述反应热后,将其通入蒸汽发生器中,并与用于转化为蒸汽的水进行热交换,从而产生蒸汽;将和所述用于转化为蒸汽的水进行了热交换的冷却剂返回至所述光气合成反应器的冷却剂流通空间,用于继续吸收合成光气产生的反应热;将所述蒸汽供应至用于蒸发浓缩盐水的盐水蒸发浓缩单元,所述蒸汽用作盐水蒸发浓缩所需的热源。一些优选实施方式中,将经盐水蒸发浓缩单元蒸发浓缩得到浓盐水送至氯碱电解池,经电解获得氯气。本发明提供的光气制备与能源综合利用的方法中,采用本发明方法得到的催化剂,为一种表面形成有磷酸铬锡和硅酸铬锡与ni通过-o-ni-o-链桥横向和纵向键合作用下形成的聚合薄层并经超声波改性固化的活性炭催化剂,提高了活性炭的耐高温和抗氧化性能;优选方案中,同时通过使用优选氧含量为10-20mg/nm3的氯气和一氧化碳,极大地降低催化剂与原料氯气及原料气中微量氧气的反应速率,明显减缓或消除活性炭高温烧蚀或粉化情况,提高催化剂的使用寿命。同时生产低游离氯、低四氯化碳的高品质光气;将光气制备工艺与蒸汽发生装置及盐水蒸发浓缩工艺相耦合,改善的光气生产装置安全稳定运行水平,使得在生产光气的同时能稳定副产蒸汽;副产蒸汽作为热源用于蒸发盐水(例如二苯基甲烷二胺/多苯基甲烷多胺或二苯基甲烷二异氰酸酯/多苯基甲烷多异氰酸酯生产过程中产生的废盐水),盐水经浓缩后可送往氯碱电解池电解生产氯气,实现能源综合利用一体化。冷却剂可以为氯苯、邻二氯苯、四氯化碳、十氢化萘或烷基苯型导热油中的一种或至少两种,优选包括邻二氯苯、二甲苯、四氯化碳、十氢化萘中的一种或至少两种,进一步优选为邻二氯苯和/或十氢化萘,更优选为十氢化萘。一些实施方式中,将所述光气合成反应器内生成的光气送入光气合成保护器中,光气合成保护器为本领域技术人员所熟知的设备。光气合成保护器内散堆装填有上文所述的催化剂(即在光气合成反应器内所装填的催化剂),光气合成保护器内的反应压力可以为0.1-0.5mpag,优选为0.2-0.4mpag;使未反应的氯气和一氧化碳进一步反应。优选的,所述光气合成保护器的出口温度控制在100℃以下,优选所述出口温度控制在50℃-80℃,更优选为60℃-70℃。将光气合成保护器的出口温度控制在上述优选温度条件下,可有效避免光气分解为co和氯气,有利于使光气合成保护器的出口输出的光气中的游离氯含量低于50mg/nm3。一些实施方式中,所述蒸汽发生器中产生的蒸汽的压力范围为0.2-1.6mpag(例如0.2mpag、0.5mpag、1.0mpag、1.3mpag、1.6mpag),优选为1.0-1.6mpag的高品质蒸汽。一些实施方式中,用于在所述盐水蒸发浓缩单元中进行蒸发浓缩的盐水来源于二苯基甲烷二胺、多苯基甲烷多胺、二苯基甲烷二异氰酸酯或多苯基甲烷多异氰酸酯生产过程中产生的废盐水,优选该盐水的氯化钠质量浓度为5%-23%,toc(总有机碳)含量为2-15ppm,利于提高盐水蒸发浓缩单元运行和经济效益,延长电解单元离子膜使用寿命。一些实施方式中,所述盐水蒸发浓缩单元采用本领域技术人员所熟知的双效或多效蒸发,优选双效或三效蒸发;经所述盐水蒸发浓缩单元蒸发浓缩得到的所述浓盐水中,氯化钠的浓度控制为300g/l-310g/l,可直接送往氯碱电解池电解生产氯气。一些实施方式中,所述光气合成反应器为列管式反应管、螺旋管式反应器、固定床列管式反应器、双管板式固定床反应器,优选本领域内广泛使用的固定床列管式反应器。具体的,光气合成反应器内,催化剂装填于列管中,冷却剂流通空间位于光气合成反应器的壳侧(光气合成反应器内围绕列管的空间)。光气合成反应器列管直径可以为25mm-70mm,优选30mm-50mm,列管长度可以为2500mm-7000mm,优选3500mm-6000mm。一些具体实施方式中,一氧化碳和氯气提前在混合气中充分混合,之后从光气合成反应器的底部进入。一氧化碳和氯气的混合方式可以为管道混合、喷嘴混合、搅拌混合、静态混合器混合等,优选为搅拌混合和静态混合器混合,更优选为静态混合器混合。氯气与一氧化碳摩尔比例可以为0.8-1.0,优选为0.85-0.98,更优选为0.90-0.95。本发明另一方面还提供一种光气制备与能源综合利用的系统,包括:光气制备单元,包括光气合成反应器,用于使氯气和一氧化碳与上文所述的催化剂接触并在催化剂作用下合成光气;所述光气合成反应器设有冷却剂流通空间,供用于吸收所述合成光气反应器内所产生的反应热的冷却剂流通;蒸汽产生单元,包括与所述冷却剂流通空间相连通的蒸汽发生器,用于接收吸收了所述反应热的冷却剂,并使其与用于转化为蒸汽的水进行热交换,从而产生蒸汽;所述光气合成反应器的冷却剂流通空间还用于接收与所述用于转化为蒸汽的水进行了热交换的冷却剂,即冷却剂在光气合成反应器的冷却剂流通空间和蒸汽发生器之间循环流动;盐水蒸发浓缩单元,与所述蒸汽产生单元连接以接收所述蒸汽产生单元产生的蒸汽,所述盐水蒸发浓缩单元用于以所述蒸汽为热源将盐水蒸发浓缩为浓盐水。一些优选实施方式中,还包括电解单元,所述电解单元包括氯碱电解池,用于接收所述浓盐水或由所述浓盐水结晶产生的干盐,并电解所述浓盐水或所述干盐以获得氯气;在一些实施方式中,所述浓盐水经结晶形成干盐后,再进入氯碱电解池电解。一些实施方式中,所述光气制备单元还包括与所述光气合成反应器的光气出口连通的光气合成保护器,用于接收所述光气出口输出的光气并使其中未反应的氯气和一氧化碳进一步反应。一些具体实施方式中,所述光气制备单元还包括与所述光气合成反应器连通的混合器,用于将氯气和一氧化碳进行混合以获得待供应至所述光气合成反应器的混合气。一些具体实施方式中,所述蒸汽产生单元还包括气包和锅炉水输送管,所述气包的蒸汽入口与所述蒸汽发生器的蒸汽出口连通,所述气包的冷凝水出口与所述蒸汽发生器的入水口连通,所述蒸汽发生器的入水口还与所述锅炉水输送管连通;所述冷凝水和锅炉水作为用于转化为蒸汽的水进入蒸汽发生器中与来自光气合成反应器的冷却剂进行热交换。具体的,所述气包的蒸汽出口与所述盐水蒸发浓缩单元连接,从而向盐水蒸发浓缩单元供应蒸汽以提供热源。此处提及的光气制备与能源综合利用的系统特别适用于实施上文所述的光气制备与能源综合利用的方法。在上文所述的光气制备与能源综合利用的方法中提到的优选特征,也同样适用于此处的光气制备与能源综合利用的系统,在此不再赘述。本发明提供的技术方案具有如下有益效果:本发明的制备方法,能提高活性炭的耐高温和抗氧化性能,制得的催化剂具有机械强度高、导热性能好、不易粉化、耐高温、抗氧化性能强的特点。本发明提供的新型改性活性炭催化剂抗氧化性能强,高温下极大降低了活性炭与氯气以及原料气中微量氧气的反应速率,利于降低光气合成保护器出口光气中四氯化碳的含量(例如低于50mg/nm3),提升产品光气质量。而在应用与光气制备时,优选将原料氯气和一氧化碳的氧含量控制为10-20mg/nm3,能极大地降低催化剂与原料氯气及原料气中微量氧气的反应速率,明显减缓或消除活性炭高温烧蚀或粉化情况,提高催化剂的使用寿命和装置安全稳定运行水平;生产低游离氯、低四氯化碳的高品质光气。本发明将基于本发明催化剂的光气生产工艺与蒸汽蒸发和盐水蒸发浓缩工艺相耦合,由于本发明催化剂的优异特性,能实现长周期运行,从而能利用光气生产中产生的反应热稳定副产蒸汽,进而能将该蒸汽作为热源供应于盐水蒸发浓缩工艺中,使废盐水得到蒸发浓缩,得到的浓盐水可结晶成干盐或直接用于电解生产氯气;因而本发明能循环利用光气合成的反应热,解决能源浪费和废盐水排放资源浪费的问题,实现能源综合利用一体化的目的。附图说明图1是一种实施方案中光气制备与能源综合利用的工艺流程图,图2:实施例5的热重表征结果(空气氛围);图3-4:分别为实施例3的改性活性炭和未改性椰壳活性炭的sem表征结果;图5:为未改性椰壳活性炭和实施例3的改性活性炭的xrd谱图。图1中,1:氯气进料管线;2:一氧化碳进料管线;3:混合器;4:光气合成反应器;5:蒸汽发生器;6:气包;7:光气合成保护器;8:冷却剂输入管线;9:锅炉水输送管;10:光气输出管线;11:一级盐水蒸发器;12:一级盐水闪蒸器;13:一级盐水泵;14:废盐水输入管线;15:二级盐水蒸发器;16:二级盐水闪蒸器;17:二级盐水泵;18:三级盐水蒸发器;19:三级盐水闪蒸器;20:三级盐水泵;21:冷凝器;22:真空泵入口气管线;23:结晶器;24:干盐输出管线;25-29:管线;30:冷却剂流通空间;31:列管。具体实施方式为了更好的理解本发明的技术方案,下面结合实施例进一步阐述本发明的内容,但本发明的内容并不仅仅局限于以下实施例。以下实施例中所用到的试验方法若未特别说明,均为本领域技术人员所掌握的常规方法;所涉及的设备和试剂,若未特别说明,均为本领域常规设备和试剂。实施例和对比例中涉及到的主要原料来源如下:一氧化碳:宁波万华工业园内造气装置生产,工业品;氯气:宁波万华氯碱公司氯氢车间生产,工业品;椰壳活性炭:日本鹤见有限公司生产,工业品;木质活性炭:福建鑫森炭业有限公司生产,工业品。光气中的游离氯含量的检测方法:(1)检测原理:cl2+2ki→2kcl+i2i2+2na2s2o3→2nai+na2s4o6(2)检测过程:配置足够的ki溶液,光气取样瓶冷冻后与玻璃漏斗相连,玻璃漏斗里加入ki溶液,打开中间的连接阀门让ki溶液充分吸收光气中的游离氯,再用准备好的na2s2o3标准溶液滴定,直至ki溶液由紫红色变为无色为止。(3)光气中的游离氯的计算方法其中,cl为光气中游离氯的含量,mg/nm3v1为滴定消耗的na2s2o3标准溶液的体积,ml;v2为气体取样瓶的体积,l;c为na2s2o3标准溶液的实际浓度,mol/l。光气中的四氯化碳含量的检测方法:光气中的四氯化碳含量采用气相色谱仪在线检测,气相色谱仪采用hewlettpackardhp5890型号,色谱柱为restaktmrtx-1crossbond100%二甲基聚硅氧烷,气相色谱升温条件为50℃维持10min,随后以15℃/min速率升温至200℃。催化剂空气氛围下失重性能测试催化剂空气氛围下的失重性能采用热重分析仪测试,热重分析仪采用mettlertoledotga-2型号,测试未改性活性炭和改性活性炭在空气氛围下的重量失重,操作条件为:升温速率20℃/min,起始温度为30℃,终止温度为600℃,天平室通入用于保护天平的保护气(氮气),该保护气流量为20ml/min,提供至催化剂的载气(即空气)流量50ml/min。实施例1制备新型改性活性炭催化剂准确称取100kg工业级磷酸,将其缓慢溶解于1t水中,搅拌溶解完全,缓慢加入28.6kg工业级硅酸,搅拌均匀后制得改性溶液。称取1t粒径为3mm的椰壳活性炭,将其浸泡在改性溶液中10h,并进行搅拌,搅拌速率控制为30r/min;浸泡后缓慢加入二甲基二氯化锡粉末44kg(0.2mol/l),加入氧化铬粉末30.4kg(0.2mol/l),反应2h后,加入粒径为0.5μm的经过450℃处理2h的氧化镍细粉15kg(0.2mol/l)反应2h,外部进行超声震荡,超声波脉冲频率控制为15khz,脉冲宽度为150ms,超声震荡时间与加入氧化镍细粉进行反应的时间一致,制得预改性活性炭。之后,取出预改性活性炭,在150℃烘箱中烘干4h,后置于高温炉中600℃加热焙烧8h,高温炉采用纯氮气保护,制得新型改性活性炭催化剂。实施例2准确称取150kg工业级磷酸,将其缓慢溶解于1t水中,搅拌溶解完全,缓慢加入75kg工业级硅酸,搅拌均匀后制得改性溶液。称取1t粒径为5mm的椰壳活性炭,将其浸泡在改性溶液中15h,并进行搅拌,搅拌速率控制为40r/min;浸泡后缓慢加入二甲基二氯化锡粉末440kg(2.0mol/l),加入氧化铬粉末456kg(3.0mol/l),反应3h后,加入粒径为0.8μm的经过500℃处理3h的氧化镍细粉112.5kg(1.2mol/l)反应3h,外部进行超声震荡,超声波脉冲频率控制为20khz,脉冲宽度为300ms,超声震荡时间与加入氧化镍细粉进行反应的时间一致,制得预改性活性炭。之后,取出预改性活性炭,在150℃烘箱中烘干8h,后置于高温炉中700℃加热焙烧12h,高温炉采用纯氮气保护,制得新型改性活性炭催化剂。实施例3准确称取125kg工业级磷酸,将其缓慢溶解于1t水中,搅拌溶解完全,缓慢加入45kg工业级硅酸,搅拌均匀后制得改性溶液。称取1t粒径为4mm的椰壳活性炭,将其浸泡在改性溶液中12h,并进行搅拌,搅拌速率控制为35r/min;浸泡后缓慢加入二甲基二氯化锡粉末165kg(0.75mol/l),加入氧化铬粉末180kg(1.2mol/l),反应2.5h后,加入粒径为0.65μm的经过500℃处理2.5h的氧化镍细粉67.5kg(0.9mol/l)反应2.5h,外部进行超声震荡,超声波脉冲频率控制为17.5khz,脉冲宽度为220ms,超声震荡时间与加入氧化镍细粉进行反应的时间一致,制得预改性活性炭。之后,取出预改性活性炭,在150℃烘箱中烘干6h,后置于高温炉中650℃加热焙烧10h,高温炉采用纯氮气保护,制得新型改性活性炭催化剂。实施例4准确称取125kg工业级磷酸,将其缓慢溶解于1t水中,搅拌溶解完全,缓慢加入45kg工业级硅酸,搅拌均匀后制得改性溶液。称取1t粒径为4mm的木质活性炭,将其浸泡在改性溶液中12h,并进行搅拌,搅拌速率控制为35r/min;浸泡后缓慢加入二甲基二氯化锡粉末165kg(0.75mol/l),加入氧化铬粉末180kg(1.2mol/l),反应2.5h后,加入粒径为0.65μm的经过500℃处理2.5h的氧化镍细粉67.5kg(0.9mol/l)反应2.5h,外部进行超声震荡,超声波脉冲频率控制为17.5khz,脉冲宽度为220ms,超声震荡时间与加入氧化镍细粉进行反应的时间一致,制得预改性活性炭。之后,取出预改性活性炭,在150℃烘箱中烘干6h,后置于高温炉中650℃加热焙烧10h,高温炉采用纯氮气保护,制得新型改性活性炭催化剂。所述实施例1-4均重复制备5次,以保证制备的新型改性活性炭催化剂重量为5t。实施例5将实施例3制得的新型改性活性炭催化剂和未经改性的粒径为4mm的椰壳活性炭进行热重表征,测试空气氛围下的失重性能,热重分析仪操作条件为:升温速率20℃/min,起始温度为30℃,终止温度为600℃,天平室通入用于保护天平的保护气(氮气)流量20ml/min,提供至催化剂的载气(即空气)流量50ml/min。实施例3制得的新型改性活性炭催化剂和未经改性的4mm椰壳活性炭热重失重谱图如图2所示。热重升温程序如下:阶段开始温度(℃)结束温度(℃)升温速率(℃/min)时间(min)130200208.52200200-30320040020104400400-3054005002056500500-307500550202.58550550-309550600202.510600600-30由图2可知,空气氛围下,未改性的4mm椰壳活性炭450℃出现明显失重,520℃失重急剧增加,实施例3制得的新型改性活性炭550℃才出现明显失重,其耐高温和抗氧化性能明显优于未改性活性炭。实施例1-2和实施例4的新型改性活性炭在进行热重表征时,也表现出与实施例3类似的结果。实施例6将实施例3制得的新型改性活性炭催化剂和未经改性的粒径4mm的椰壳活性炭进行sem表征,测试二者的活性炭表面膜层形貌、膜层厚度和膜层的连续性,试验采用hitachi的tm-1000型扫描电镜。实施例3制得的新型改性活性炭催化剂和未经改性的椰壳活性炭的sem扫描图谱分别如图3、4所示。由图3可知,实施例3制得的新型改性活性炭催化剂表面覆盖一层致密的连续薄层,膜层厚度为7μm左右;而图4的未改性的4mm椰壳活性炭表面无此特征,且活性炭表面有明显裂纹。实施例1-2和实施例4的新型改性活性炭在进行sem表征时,结果与实施例3类似。实施例7将实施例3制得的新型改性活性炭催化剂和未经改性的粒径为4mm的椰壳活性炭进行xrd表征,x射线衍射仪为日本理学株式会社的d/max2200型,衍射条件为:cukα,波长0.1541nm,电压40kv,扫描速度3.5°·min-1,电流40ma。未经改性的4mm椰壳活性炭和实施例3制得的新型改性活性炭催化剂的xrd谱图分别如图5所示。由图5可知,相比未改性的椰壳活性炭,实施例3制得的新型改性活性炭催化剂在2θ为43°出现了较为明显的nio特征衍射峰,在2θ为28.5°出现了crpo4特征衍射峰,在2θ为35°出现了sn2p2o7特征衍射峰,在2θ为33.5°和37.5°出现了cr2sio5和sn2sio4特征衍射峰,在2θ为52.5°出现了ni2p2o7特征衍射峰,在2θ为56°出现了ni2sio4特征衍射峰,实施例1-2和实施例4的新型改性活性炭在进行xrd表征时,也表现出与实施例3类似的结果。以下实施例为光气制备与能源综合利用的实施例,以下实施例中所使用的光气制备与能源综合利用的系统介绍如下:参见图1,主要包括光气制备单元100、蒸汽产生单元200和盐水蒸发浓缩单元300。其中,光气制备单元100包括混合器3、光气合成反应器4和光气合成保护器7。混合器3连接有氯气进料管线1和一氧化碳进料管线2,用于将原料氯气和原料一氧化碳混合。一氧化碳和氯气的混合方式可以为管道混合、喷嘴混合、搅拌混合、静态混合器混合等。混合器3与光气合成反应器4连通,以将氯气和一氧化碳的混合气输入光气合成反应器4。光气合成反应器4可以为列管式反应器、螺旋管式反应器、固定床列管式反应器或双管板式固定床反应器,在以下实施例中具体采用的是固定床列管式反应器。光气合成反应器列管直径可以为25mm-70mm,优选30mm-50mm,列管长度可以为2500mm-7000mm,优选3500mm-6000mm。光气合成保护器7与光气合成反应器4的光气出口连通,用于接收光气合成反应器的光气出口输出的光气,并使其中未反应的氯气和一氧化碳进一步反应完全。混合器3输出的混合气由光气合成反应器4的底部进入。光气合成反应器4的列管31内装填催化剂,光气合成反应器4的冷却剂流通空间30(即壳侧)内流通冷却剂,冷却剂用于吸收在光气合成过程中产生的反应热;冷却剂可以是氯苯、邻二氯苯、四氯化碳、十氢化萘或烷基苯型导热油中的一种或至少两种,优选包括邻二氯苯、二甲苯、四氯化碳、十氢化萘中的一种或至少两种,进一步优选为邻二氯苯或十氢化萘,更优选为十氢化萘。蒸汽产生单元200包括蒸汽发生器5和气包6,其中,蒸汽发生器5的冷却剂入口(图中未示出)与光气合成反应器4的壳侧的冷却剂出口(图中未示出)通过管线25连通,蒸汽发生器5的冷却剂出口(图中未示出)与光气合成反应器4的壳侧的冷却剂入口(图中未示出)通过管线26连通。光气合成反应器4的壳侧的冷却剂入口还与用于供应冷却剂的冷却剂输入管线8连通。气包6的蒸汽入口(图中未示出)与蒸汽发生器5的蒸汽出口(图中未示出)通过管线27连通,气包6的冷凝水出口(图中未示出)与蒸汽发生器5的入水口(图中未示出)通过管线28连通;蒸汽发生器5的入水口还与锅炉水输送管9连通。吸收了光气合成反应器4内的反应热的冷却剂通过管线25通入至蒸汽发生器5内,与来自气包6的冷凝水/来自锅炉水输送管9的锅炉水进行热交换,从而产生蒸汽;热交换后降温的冷却剂通过管线26循环至光气合成反应器4中。具体的,光气合成反应器4的壳侧的冷却剂出口位于上部,而光气合成反应器4的壳侧的冷却剂入口位于下部,反应器壳侧上部设置有气相蒸发空间,冷却剂移除反应热的方式为带走气化潜热,冷却剂气化后进入蒸汽发生器5进行换热。盐水蒸发浓缩单元300与蒸汽产生单元200连接以接收蒸汽产生单元产生的蒸汽,盐水蒸发浓缩单元300用于以所述蒸汽为热源将盐水蒸发浓缩为浓盐水。具体的,盐水蒸发浓缩单元300采用本领域技术人员所熟知的双效或多效蒸发,优选双效或三效蒸发,具体的蒸发工艺或所依据的设备均为本领域技术人员所熟知的。在以下实施例中采用三效蒸发,具体可参见图1,下面结合图1进行示例介绍:具体包括依次串联的一级浓缩单元301、二级浓缩单元302和三级浓缩单元303,各个浓缩单元均包括盐水蒸发器(11、15或18)、盐水闪蒸器(12、16或19)和盐水泵(13、17或20)。气包6中的蒸汽经管线29送入一级浓缩单元301的一级盐水蒸发器11中,经废盐水输入管线14进入一级盐水闪蒸器12的废盐水经一级盐水泵13送至一级盐水蒸发器11,与来自气包6的蒸汽热交换,之后再进入一级盐水闪蒸器12闪蒸浓缩;在泵13的作用下,经闪蒸浓缩得到的一级浓盐水被送入二级浓缩单元302的二级盐水闪蒸器16,在二级浓缩单元302中的具体处理过程与一级浓缩单元301相同;经二级浓缩单元302闪蒸浓缩后,再进入三级浓缩单元303进行与一级浓缩单元301和二级浓缩单元302相同的闪蒸浓缩处理。而一级浓缩单元301中的一级盐水闪蒸器12中的蒸汽流入二级浓缩单元302的二级盐水蒸发器15中,用于与盐水热交换;同样的,二级浓缩单元302中的二级盐水闪蒸器16中的蒸汽流入三级浓缩单元303的三级盐水蒸发器18中,用于与盐水热交换。三级盐水闪蒸器19中排出的蒸汽进入冷凝器21中冷凝。而经三级浓缩单元303闪蒸浓缩得到的浓盐水可进一步进入结晶器23中结晶,得到干盐;或者,将得到的浓盐水中的氯化钠的浓度控制为300g/l-310g/l,可直接送往下游的氯碱电解池电解生产氯气。实施例8实施例1制得的新型改性活性炭催化剂装填于光气合成反应器和光气合成保护器中,反应器列管直径为40mm,长度为5000mm。光气合成反应器中装填催化剂共4.8t,氯气与一氧化碳中总的氧含量为15mg/nm3,氯气和一氧化碳以摩尔比0.93在静态混合器中混合后进入光气合成反应器反应生成光气,光气合成反应器压力控制为0.4mpag,光气合成反应器内的入口温度为30℃,光气合成反应器的光气出口的温度控制在210℃,光气合成反应器列管中带有热电偶监控热点温度;光气合成反应器出口光气进入光气合成保护器,未反应的氯气和一氧化碳进一步反应完全,光气合成保护器压力控制为0.4mpag,光气合成保护器出口温度控制在65℃。光气合成反应器壳侧冷却剂为十氢化萘,十氢化萘在壳侧气化后进入蒸汽发生器与锅炉水(还有部分来自气包的冷凝水)换热,副产蒸汽压力为1.0mpag,蒸汽流量为21t/h,蒸汽用于三效蒸发废盐水,废盐水具体来自二苯基甲烷二胺/多苯基甲烷多胺或二苯基甲烷二异氰酸酯/多苯基甲烷多异氰酸酯生产过程中产生的废盐水,废盐水中氯化钠质量浓度为15%,toc含量为10ppm。废盐水经浓缩后氯化钠浓度为309g/l,浓盐水送往氯碱电解池电解生产氯气,实现能源与三废的综合利用。光气合成保护器出口的光气中的游离氯和四氯化碳分析结果如表1所示。实施例9-11实施例9-11中光气的制备方法和实施例8在相同条件下进行,不同之处仅在于光气合成反应器中装填的新型改性活性炭催化剂,实施例9、10、11所用活性炭催化剂分别对应于实施例2、3、4。具体分析结果如表1所示。实施例12实施例3制得的新型改性活性炭催化剂装填于光气合成反应器和光气合成保护器中,反应器列管直径为40mm,长度为5000mm。光气合成反应器中装填催化剂共4.8t,氯气与一氧化碳中总的氧含量为50mg/nm3,氯气和一氧化碳以摩尔比0.93在静态混合器中混合后进入光气合成反应器反应生成光气,光气合成反应器压力控制为0.4mpag,光气合成反应器内的入口温度为30℃,光气合成反应器的光气出口的温度控制在210℃,光气合成反应器列管中带有热电偶监控热点温度;光气合成反应器出口光气进入光气合成保护器,未反应的氯气和一氧化碳进一步反应完全,光气合成保护器压力控制为0.4mpag,光气合成保护器出口温度控制在65℃。光气合成反应器壳侧冷却剂为十氢化萘,十氢化萘在壳侧气化后进入蒸汽发生器与锅炉水(还有部分来自气包的冷凝水)换热,副产蒸汽压力为1.0mpag,蒸汽流量为21t/h,蒸汽用于三效蒸发废盐水,废盐水具体来自二苯基甲烷二胺/多苯基甲烷多胺或二苯基甲烷二异氰酸酯/多苯基甲烷多异氰酸酯生产过程中产生的废盐水,废盐水中氯化钠质量浓度为15%,toc含量为10ppm。废盐水经浓缩后氯化钠浓度为309g/l,浓盐水送往氯碱电解池电解生产氯气,实现能源与三废的综合利用。光气合成保护器出口的合成光气中的游离氯和四氯化碳分析结果如表1所示。实施例13实施例3制得的新型改性活性炭催化剂装填于光气合成反应器和光气合成保护器中,反应器列管直径为40mm,长度为5000mm。光气合成反应器中装填催化剂共4.8t,氯气与一氧化碳中总的氧含量为15mg/nm3,氯气和一氧化碳以摩尔比0.93在静态混合器中混合后进入光气合成反应器反应生成光气,光气合成反应器压力控制为0.4mpag,光气合成反应器内的入口温度为30℃,光气合成反应器的光气出口的温度控制在210℃,光气合成反应器列管中带有热电偶监控热点温度;光气合成反应器出口光气进入光气合成保护器,未反应的氯气和一氧化碳进一步反应完全,光气合成保护器压力控制为0.4mpag,光气合成保护器出口温度控制在65℃。光气合成反应器壳侧冷却剂为邻二氯苯,邻二氯苯在壳侧气化后进入蒸汽发生器与锅炉水(还有部分来自气包的冷凝水)换热,副产蒸汽压力为1.0mpag,蒸汽流量为21t/h,蒸汽用于三效蒸发废盐水,废盐水具体来自二苯基甲烷二胺/多苯基甲烷多胺或二苯基甲烷二异氰酸酯/多苯基甲烷多异氰酸酯生产过程中产生的废盐水,废盐水中氯化钠质量浓度为15%,toc含量为10ppm。废盐水经浓缩后氯化钠浓度为309g/l,浓盐水送往氯碱电解池电解生产氯气,实现能源与三废的综合利用。光气合成保护器出口的合成光气中的游离氯和四氯化碳分析结果如表1所示。对比例1将未经改性的4mm椰壳活性炭催化剂装填于光气合成反应器和光气合成保护器中,反应器列管直径为40mm,长度为5000mm。光气合成反应器中装填活性炭共4.8t,氯气与一氧化碳中总的氧含量为15mg/nm3,氯气和一氧化碳以摩尔比0.93在静态混合器中混合后进入光气合成反应器反应生成光气,光气合成反应器压力控制为0.4mpag,光气合成反应器内的入口温度为30℃,光气合成反应器的光气出口的温度控制在210℃,光气合成反应器列管中带有热电偶监控热点温度,光气合成反应器出口光气进入光气合成保护器,未反应的氯气和一氧化碳进一步反应完全,光气合成保护器压力控制为0.4mpag,光气合成保护器出口温度控制在65℃。光气合成反应器壳侧冷却剂选取为十氢化萘,十氢化萘气化后进入蒸汽发生器与锅炉水(还有部分来自气包的冷凝水)换热,副产蒸汽压力为1.0mpag,蒸汽流量为21t/h,因副产蒸汽流量不稳定,将其并入蒸汽管网。二苯基甲烷二胺/多苯基甲烷多胺或二苯基甲烷二异氰酸酯/多苯基甲烷多异氰酸酯生产过程中产生的废盐水与盐酸中和,中和后ph控制在7-9范围内,氯化钠质量浓度控制在15%-23%范围内,处理合格后排海。合成光气中的游离氯和四氯化碳分析结果如表1所示。对比例2将未经改性的4mm椰壳活性炭催化剂装填于光气合成反应器和光气合成保护器中,反应器列管直径为40mm,长度为5000mm。光气合成反应器中装填活性炭共4.8t,氯气与一氧化碳中总的氧含量为150mg/nm3,氯气和一氧化碳以摩尔比0.93在静态混合器中混合后进入光气合成反应器反应生成光气,光气合成反应器压力控制为0.4mpag,光气合成反应器内的入口温度为30℃,光气合成反应器的光气出口的温度控制在210℃,光气合成反应器列管中带有热电偶监控热点温度;光气合成反应器出口光气进入光气合成保护器,未反应的氯气和一氧化碳进一步反应完全,光气合成保护器出口温度控制在65℃。光气合成反应器壳侧冷却剂选取为十氢化萘,十氢化萘气化后进入蒸汽发生器与锅炉水(还有部分来自气包的冷凝水)换热,副产蒸汽压力为1.0mpag,蒸汽流量为21t/h,因副产蒸汽流量不稳定,将其并入蒸汽管网。二苯基甲烷二胺/多苯基甲烷多胺或二苯基甲烷二异氰酸酯/多苯基甲烷多异氰酸酯生产过程中产生的废盐水与盐酸中和,中和后ph控制在7-9范围内,氯化钠质量浓度控制在15%-23%范围内,处理合格后排海。合成光气中的游离氯和四氯化碳分析结果如表1所示。表1:实施例及对比例的性能数据注:表1中,运行周期指实际运行时长。从以上实验结果可见,基于本发明的方法得到的改性活性炭催化剂,具有更长的使用寿命,在光气制备中,装置能更持久的安全稳定运行;与蒸汽蒸发单元联合,能稳定产出高质量蒸汽,为废盐水的蒸发浓缩提供热源,达到能源综合利用,避免资源浪费。而且,基于本发明的方法得到的改性活性炭催化剂,光气合成反应器热点温度相对更低,催化剂损失量少,光气中的游离氯和四氯化碳含量低;而且在10-50mg/nm3,特别是10-20mg/nm3的原料气氧含量下,光气合成反应器热点温度更低,催化剂损失量更少,运行周期更长,光气中的游离氯和四氯化碳含量更低。同时,从本发明改性活性炭的催化剂损失量更低和具有更长的运行周期来看,该改性的催化剂具有较佳的耐高温和抗氧化性能。本领域技术人员可以理解,在本说明书的教导之下,可对本发明做出一些修改或调整。这些修改或调整也应当在本发明权利要求所限定的范围之内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1