一种锚定型催化剂制备方法和催化氧化重油脱硫、脱氮、脱金属及脱酸方法与流程
本发明属于一种重油改质加工方法。具体涉及一种将非均相选择性氧化用于重油脱硫、脱氮、脱金属及脱酸的改质加工方法
背景技术:
随着世界范围内原油资源逐步趋于重质化、劣质化,环保法规的日益严格以及汽柴油质量标准的不断升级,重质原油高效加工利用已成为当今炼油工业面临的重大挑战。重质原油加工产品轻质化和清洁化、炼制过程清洁化和低碳化,提升原油转化效率,已成为炼油企业重点关注的问题。
随着原油日益劣质化,高金属、高残炭、高硫、高氮、高酸值、高黏度是重质原油的普遍特性。重质原油加氢加工成本越来越高,固定床、悬浮床和沸腾床等加氢技术,需在高温、高压和昂贵的设备和催化剂等苛刻条件下进行,加工成本高、工艺复杂,这些工艺的推广应用受到一定程度的限制。相对加氢技术,氧化脱硫技术可在常温、常压下进行,不耗费氢气、不增加碳排放,设备投资较少,特别是对催化加氢难以脱除的稠环噻吩类含硫化合物的脱硫效率高。
cn201810661570.3公开了一种用于重油氧化脱硫的方法。具体过程如下:向含硫重油中分散通入氧气或臭氧,加热至135-150℃发生氧化脱硫反应。对于每100ml含硫重油,通入氧气的速度是300~400ml/min,上述氧化脱硫反应在油浴反应器中进行。然而氧气和臭氧氧化反应的选择性差,会造成烃类物质氧化、聚合,影响油品质量。
cn201810352278.3公开了一种化学链循环部分氧化重油加工方法,用于生产汽油和柴油。重油进入部分氧化单元与载氧体进行部分氧化反应,得部分氧化产物和还原态载氧体,氧化条件为:反应温度400~600℃;还原态的载氧体与空气或水蒸气在再生单元中400~700℃下反应实现载氧体再生,再生后的载氧体与重油进行部分氧化反应。该工艺反应温度高、工艺复杂。
目前mo基催化剂被广泛应用于各类油品的氧化脱硫,一般mo基催化剂制备采用浸渍法,操作简便,经济实用。但这种方法制备的催化剂,活性组分mo元素通常在载体表面分散程度较差,容易导致mo物种的堆积。与此同时,由于催化剂是亲水性,极性的氧化产物(砜类物质)极易吸附在催化剂表面,降低了催化剂的有效活性位点,容易失活。
技术实现要素:
针对负载型mo基催化剂活性低,易失活的缺点,本发明提供一种锚定型催化剂制备方法和催化氧化重油脱硫、脱氮、脱金属及脱酸方法。该催化剂能在常压温和条件下,以h2o2、有机过氧化氢为氧化剂,催化重油中的硫化物、氮化物和金属螯合物反应,氧化后的重油经过碱洗,可以实现脱硫、脱氮、脱金属和脱酸。
为解决上述技术问题,本发明采用如下技术方案:
本发明提供一种锚定型催化剂制备方法,包括如下步骤:
(1)sio2与无水乙醇混合,搅拌条件下缓慢加入硅烷化试剂,100℃回流48h。冷却至室温后,将溶液离心分离出固体,然后用无水乙醇洗涤三次,100℃干燥12h,得到改性的sio2。
(2)取一定质量的mo的前驱体加入无水乙醇,然后加入一定量的改性sio2,搅拌,80~100℃回流24h,冷却至室温,将溶液离心分离固体催化剂,然后用无水乙醇洗涤催化剂3次,80℃干燥12h,得到锚定型催化剂。
优选地,在步骤1中,硅烷试剂包括但不仅限于n-(2-氨乙基)-3-氨丙基三甲氧基硅烷,γ-巯丙基三甲氧基硅烷和γ-氨丙基三乙氧基硅烷。
优选地,在步骤2中,mo的前驱体为钼酸铵、磷钼酸。
上述方法制备的锚定型催化剂催化氧化重油脱硫、脱氮、脱金属及脱酸方法,包括以下步骤:
s1.重油加入稀释剂稀释后,与氧化剂进入氧化单元与催化剂接触,进行选择性氧化反应,得到氧化重油,其中氧化条件为:反应温度为60~90℃,稀释剂与重油的体积比为1~2,反应停留时间为20~60分钟;
s2.步骤s1得到的氧化重油与浓度为0.5~2mol/l的naoh溶液一起进入碱洗单元,在温度200~300℃下,进行碱洗,碱洗时间为2~4小时。
优选地,所述氧化剂选自过氧化氢、叔丁基过氧化氢或过氧化羟基异丙苯中的一种及几种。
优选地,所述重质油选自重质原油、含酸原油、超稠油、常压渣油、减压渣油、减压蜡油、煤焦油,煤液化残渣油、页岩油、油砂沥青,或其他二次加工馏分油中的的一种或几种混合物。同时,本发明制备的锚定型催化剂对汽油、柴油和船用燃料油等馏分油不用加入稀释剂同样具有很好的脱硫、脱氮、脱金属及脱酸效果。
优选地,在步骤s1中,加入稀释剂对重油进行稀释,可以降低重油粘度,所述的稀释剂为正己烷、苯、甲苯、二甲苯、异丙苯中的一种或几种溶剂的混合物。这些溶剂本身就可以作为油品组分,增加油品收率,同时不会对油品质量造成影响。
优选地,在步骤s1中,氧化单元为固定床、流化床或釜式反应器。
优选地,在步骤s2中,碱洗单元为釜式反应器或塔式反应器。
与现有技术相比,本发明具有以下有益效果:
(1)本发明提供一种锚定型催化剂制备方法,该催化剂能在常压温和条件下,以h2o2、有机过氧化氢为氧化剂,催化重油中的硫化物、氮化物和金属螯合物反应,氧化后的重油经过碱洗,可以实现脱硫、脱氮、脱金属和脱酸,锚定型催化剂活性组分mo的分散度和催化活性高于普通负载型催化剂。
(2)本发明制备的锚定型催化剂中,活性组分mo元素通过有机基团锚定到sio2表面,mo元素分散好,同时sio2表面被有机基团改性后,增加了疏水性,反应极性产物不容易被吸附在sio2表面,催化剂活性高、寿命长。表面疏水性增强,氧化产物砜类物质不易被吸附,催化剂寿命远远高于普通负载型mo基催化剂。
(3)本发明锚定型催化剂制备过程中,碱洗过程中的碱液可以循环使用,无废液排放。
(4)本发明制备的锚定型催化剂可以在温和条件下实现连续化重油脱硫、脱氮、脱金属和脱酸。
(5)本发明工艺简单、可以在温和条件下实现对重油高效改质。
附图说明
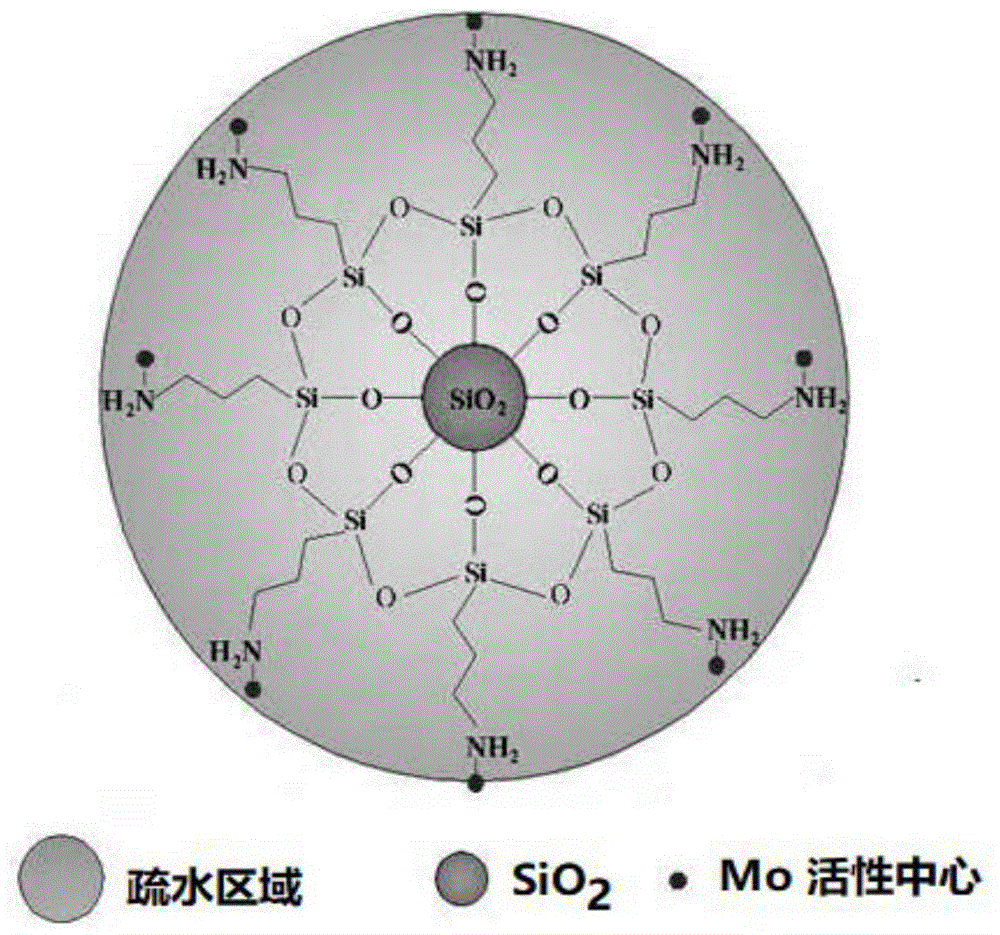
图1为实施例1和实施例2所制备的锚定型催化剂结构示意图。
图2为实施例1所制备的锚定型催化剂与普通负载型mo/sio2的透射电镜扫描对比图。
图3为实施例1所制备的锚定型催化剂和普通负载型mo/sio2催化活性和使用寿命比较图。
图4为实施例1所制备的锚定型催化剂和普通负载型mo/sio2催化氧化重油前后的红外光谱图。
图5为本发明制备的锚定型催化剂催化氧化重油脱硫、脱氮、脱金属及脱酸的流程示意图。
具体实施方式
下面通过具体实施例对本发明作进一步详述,以下实施例只是描述性的,不是限定性的,不能以此限定本发明的保护范围。如无特殊说明,本发明所采用的实验设备、材料和试剂均可从商业途径获得。下述实施例中涉及的氧化单元反应器以固定床反应器为例,碱洗单元为釜式反应器为例来更清楚阐述本发明,需要说明的是,氧化单元采用流化床或釜式反应器,碱洗单元采用塔式反应器均可以达到本发明的效果,在此不一一赘述。
具体技术流程如图5所示,重油和氧化剂混合进入氧化单元,在氧化单元中与锚定型催化剂接触反应,氧化后重油与碱液进入碱洗单元,在碱洗单元内重油与碱液接触反应,反应后得到脱硫、脱氮、脱金属和脱酸后的重油,碱液分离后可以循环使用。
实施例1~3为催化剂制备实施例,实施例4~9为本方法实施例。
实施例1
取6.0gsio2与120ml无水乙醇混合,搅拌条件下缓慢加入4.5gγ-氨丙基三乙氧基硅烷,100℃回流48h。冷却至室温后,将溶液离心分离出固体,然后用无水乙醇洗涤固体三次,100℃干燥12h,得到改性的sio2。
取0.4gh3po4·12moo3加入180ml无水乙醇中,待h3po4·12moo3完全溶解之后,加入3.0g改性的sio2,80~100℃回流24h。反应结束后,冷却至室温,将溶液离心分离的固体用无水乙醇洗涤3次,80℃干燥12h,得到锚定型催化剂。
实施例2
取5.0gsio2与120ml无水乙醇混合,搅拌条件下缓慢加入5gn-(2-氨乙基)-3-氨丙基三甲氧基硅烷,100℃回流48h。冷却至室温后,将溶液离心取出固体,然后用无水乙醇洗涤三次,100℃干燥12h,得到改性的sio2。
取0.3g钼酸铵加入180ml无水乙醇中,待钼酸铵完全溶解之后,加入3.0g改性的sio2,80~100℃回流24h。反应结束后,冷却至室温,将溶液离心分离的固体用无水乙醇洗涤3次,80℃干燥12h,得到锚定型催化剂。
实施例3
取5.0gsio2与120ml无水乙醇混合,搅拌条件下缓慢加入4gγ-巯丙基三甲氧基硅烷,100℃回流48h。冷却至室温后,将溶液离心取出固体,然后用无水乙醇洗涤三次,100℃干燥12h,得到改性的sio2。
取0.5gh3po4·12moo3加入180ml无水乙醇中,待钼酸铵完全溶解之后,加入3.0g改性的sio2,80~100℃回流24h。反应结束后,冷却至室温,将溶液离心分离的固体用无水乙醇洗涤3次,80℃干燥12h,得到锚定型催化剂。
图1为实施例1和实施例2制备的锚定型催化剂结构示意图,从图中可以看出,活性组分mo元素通过有机基团联接到sio2表面,mo元素分散非常好,这是其反应活性高的主要原因。同时sio2表面被有机基团改性后,增加的疏水性,在催化剂周围形成一个疏水区域,反应极性产物(如砜类物质)不容易被吸附在sio2表面,这是其使用寿命长的主要原因。
图2为实施例1制备的锚定型催化剂与普通负载型催化剂的透射电镜扫描图,通过图2(a)和图2(b)可以直观的看出,与普通负载型mo/sio2催化剂相比,锚定型催化剂中mo物种具有更好的分散度。锚定型催化剂上mo物种颗粒尺寸较小且分布均匀,颗粒粒径统计结果(图2(c)和图2(d))显示,锚定型催化剂上mo物种颗粒的平均大小在3.1±0.05nm,而普通负载型mo/sio2催化剂上mo物种的颗粒较大,颗粒平均尺寸在9.5±1.5nm。
图3为在相同条件下普通负载型mo/sio2和锚定型催化剂催化异丙基过氧化氢氧化重质原油催化活性和使用寿命比较。从图3中可以看出,在相同条件下锚定型催化剂的催化活性高于普通负载型mo/sio2。锚定型催化剂在使用10次后活性只有微小的降低,而普通负载型mo/sio2活性随使用次数的增加下降十分显著,这说明锚定型催化剂的活性和使用寿命明显优于普通负载型mo/sio2。锚定型催化剂的催化活性高于普通负载型mo/sio2,主要归功于活性组分mo元素在锚定型催化剂上分散度高,粒径小。
实施例4
以异丙苯为稀释剂,按体积比1:1稀释重质原油,实施例1制备的催化剂在内径为10mm的固定床反应器进重质原油氧化,反应温度为60℃,停留时间为40分钟,氧化剂叔丁基过氧化氢与原油体积配比为2:5。氧化后重质原油进入釜式反应器进行碱洗,naoh水溶液的浓度为2mol/l,碱液与重质原油体积比为2:1,温度为300℃,反应时间为3小时。反应结果如表1所示。
表1重质原油催化氧化后脱硫、脱氮和脱金属结果
实施例5
以异丙苯为稀释剂,按体积比1:1稀释重质原油,实施例2制备的催化剂在内径为10mm的固定床反应器进重质原油氧化,反应温度为80℃,停留时间为40分钟,氧化剂异丙苯基过氧化氢与原油体积配比为2:5。氧化后重质原油进入釜式反应器进行碱洗,naoh浓度为2mol/l,碱液与重质原油体积比为2:1,温度为300℃,反应时间为3小时。反应结果如表2所示。
表2重质原油催化氧化后脱硫、脱氮和脱金属结果
实施例6
以二甲苯为稀释剂,按体积比1:1稀释重质原油,实施例3制备的催化剂在内径为10mm的固定床反应器进重质原油氧化,反应温度为80℃,停留时间为40分钟,氧化剂为异丙苯基过氧化氢与原油体积配比为2:5。氧化后重质原油进入釜式反应器进行碱洗,naoh浓度为2mol/l,碱液与重质原油体积比为2:1,温度为300℃,反应时间为3小时。反应结果如表3所示。
表3重质原油催化氧化后脱硫、脱氮和脱金属结果
实施例7
以甲苯为稀释剂,按体积比2:1稀减压渣油,方案一制备的催化剂在内径为10mm的固定床反应器进减压渣油氧化,反应温度为60℃,停留时间为40分钟,氧化剂叔丁基过氧化氢与原油体积配比为2:5。氧化后减压渣油进入釜式反应器进行碱洗,naoh浓度为2mol/l,碱液与重质原油体积比为2:1,温度为300℃,反应时间为3小时。反应结果如表4所示。
表4减压渣油催化氧化后脱硫、脱氮和脱金属结果
实施例8
以二甲苯为稀释剂,按体积比1:1稀释沥青蜡油,实施例1制备的催化剂在内径为10mm的固定床反应器进减压渣油氧化,反应温度为60℃,停留时间为40分钟,氧化剂过氧化氢与原油体积配比为3:5。氧化后沥青蜡油进入釜式反应器进行碱洗,naoh浓度为2mol/l,碱液与沥青蜡油体积比为2:1,温度为300℃,反应时间为3小时。反应结果如表5所示。
表5沥青蜡油催化氧化后脱硫、脱氮和脱金属结果
实施例9
以甲苯为稀释剂,按体积比1:1稀释减压蜡油,实施例1制备的催化剂在内径为10mm的固定床反应器进减压渣油氧化,反应温度为80℃,停留时间为40分钟,氧化剂异丙苯基过氧化氢原油体积配比为2:5。氧化后减压渣油进入釜式反应器进行碱洗,naoh浓度为2mol/l,碱液与减压蜡油体积比为2:1,温度为300℃,反应时间为3小时。反应结果如表6所示。
表6减压蜡油催化氧化后脱硫、脱氮和脱金属结果
采用锚定型催化剂对不同类型的重油进行催化氧化,结果如表1到表6所示。从表1到表6可以看出,经过氧化脱硫和碱洗后,重油中的氮化物、硫化物、酸值和金属都有不同程度的降低。通过本发明制备的催化剂和催化氧化重油脱硫、脱氮、脱金属及脱酸方法,可以在常压温和条件下实现大幅降低原油中的硫化物、氮化物、酸值和金属元素。
图4分别对比了催化氧化重油反应前后锚定型催化剂和普通负载型mo/sio2的红外光谱。从图4中可以看出,使用后的mo/sio2和锚定型催化剂均保持了原催化剂的结构特征峰,然而对于使用后的mo/sio2催化剂在1288cm-1和1166cm-1存在两个不同的特征峰,分别为s=o的不对称和对称拉伸振动峰,可知其归属于二苯并噻吩砜中s=o的特征峰,说明极性产物二苯并噻吩砜易吸附在亲水性的mo/sio2催化剂表面,导致mo/sio2催化剂的活性急速下降。而对于使用后的锚定型催化剂的ft-ir光谱上没有出现类似的峰,说明产物砜类物质不易吸附在疏水性的催化剂表面,这是由于锚定型催化剂表面形成了疏水区域,对极性产物砜类物质具有排斥作用的结果。由于产物砜类物质不被吸附,因此锚定型催化剂的寿命明显高于普通负载型mo/sio2催化剂。
显然,本领域的技术人员可以对本发明进行各种改动和变型而不脱离本发明的精神和范围。这样,倘若本发明的这些修改和变型属于本发明权利要求及其等同技术的范围之内,则本发明也意图包含这些改动和变型在内。
- 还没有人留言评论。精彩留言会获得点赞!