一种三维有序大孔氧缺陷型二氧化铈催化剂及其制备方法和应用与流程
本发明属于环境催化与无机金属催化剂材料的制备领域,具体涉及一种三维有序大孔氧缺陷型二氧化铈催化剂及其制备方法和应用。
背景技术:
大量文献已证实光热催化净化挥发性有机化合物(vocs)的高效性。光热催化技术的核心是高活性和高稳定催化剂的开发。二氧化铈本身具备优良的光学性能、储氧释氧能力与热稳定性,在光热催化领域应用广泛。但在处理一些难降解vocs过程中,如苯系物、直链烷烃和环烷烃等,所需矿化温度高,大量中间产物快速形成,中间产物甚至积碳易累积在催化剂表面导致其逐渐失活,严重影响二氧化铈催化剂的催化稳定性。
目前,研究者已经尝试了多种改性方法以优化二氧化铈的催化活性与稳定性。主要集中于形貌调控、元素掺杂和贵金属负载。大量文献已报道了孔径均一、排列有序的开放孔道结构,如纳米管阵列和三维有序大孔等,能有效提高二氧化铈与反应物的接触面积,并且每个孔道可看作一个独立的反应场所,能同时限域活性组分与富集反应物分子;此外其有序的孔道结构不仅有利于热量传递和反应物质间的交换,而且能储存一定波长的入射光,减少光子快速散射,促进光热催化反应。另外,促进活性氧的生成是提高催化性能的关键,单靠形貌调控,并不能从本质上提高二氧化铈的氧化还原性能。部分研究者通过元素掺杂(如bi、n、mn、co、k)拓宽二氧化铈光响应范围以及提高其晶格氧迁移能力,或通过负载强氧化活性的贵金属(如pt、pd),提高对氧的活化能力。但由于元素掺杂会降低二氧化铈的结构稳定性,而贵金属颗粒在反应过程中也容易粗化与团聚,导致改性后的催化剂存在稳定性较差和贵金属用量大、成本高等问题。
技术实现要素:
为了解决上述现有技术中存在的不足和缺点,本发明的首要目的在于提供一种三维有序大孔氧缺陷型二氧化铈催化剂的制备方法。该方法通过调控催化剂的形貌、氧空位与酸性位,使得所制二氧化铈孔道大小均一有序、且富含氧空位与弱酸性位,有利于反应物的吸附及氧气活化,并提升其在光热催化过程的催化活性与抗积碳能力,解决了传统二氧化铈催化剂活性不高、催化稳定性差的问题。
本发明的另一目的在于提供一种上述制备方法制备得到的三维有序大孔氧缺陷型二氧化铈催化剂。
本发明的再一目的在于提供上述三维有序大孔氧缺陷型二氧化铈催化剂的应用。
本发明的目的通过下述技术方案实现:
一种三维有序大孔氧缺陷型二氧化铈催化剂的制备方法,包括以下步骤:
s1、取六水合硝酸铈与一水柠檬酸粉末,加入醇溶液,超声后得到均匀透明溶液a;
s2、将聚甲基丙烯酸甲酯(pmma)微球粉末浸渍在溶液a中,超声均匀后,抽滤除去多余液体,得到固体b,并将固体b依次进行真空干燥、煅烧i和煅烧ii操作,得到固体c;
s3、将步骤s2所得固体c移入固定床反应器,后进行煅烧iii、水蒸气处理和煅烧iv操作,得到三维有序大孔氧缺陷型二氧化铈催化剂;
步骤s3中煅烧iii与水蒸气处理同时进行,其过程为:采用氮气鼓泡装置控制反应器内湿度,控制湿度为10%~90%,同时在体积比为1:0.1~10的氢气与氮气混合气氛围下200~600℃煅烧1~24h;所述煅烧iv的过程为:在保持所述煅烧iii温度条件下通入氮气吹扫1h进行除湿处理后,在体积比为1:0.1~10的空气与氮气混合气氛围下200~600℃煅烧1~24h。
所述步骤s1中的醇溶液为乙二醇与甲醇混合液或乙醇,所述乙二醇与甲醇混合液中乙二醇和甲醇的体积比为1:0.1~10;所述六水合硝酸铈与一水柠檬酸粉末的摩尔比为1:0.1~10。
所述步骤s1中的六水合硝酸铈与一水柠檬酸的总质量,与醇溶液体积比按照0.9~1.1g/ml。
所述步骤s2中聚甲基丙烯酸甲酯微球的直径为50~500nm;所述聚甲基丙烯酸甲酯微球与溶液a按照质量体积比0.25~0.3g/ml;所述真空干燥的温度为30~80℃,时间为1~100h;所述煅烧ⅰ与煅烧ii在管式炉进行,所述煅烧i的过程为:在氩气氛围下100~1000℃煅烧1~36h,所述煅烧ii的过程为:在空气氛围下300~1000℃煅烧1~36h。
一种由上述的制备方法制备得到的三维有序大孔氧缺陷型二氧化铈催化剂。
上述的三维有序大孔氧缺陷型二氧化铈催化剂在光热催化净化苯乙烯、正己烷和环己烷中的应用。
与现有技术相比,本发明具有如下优点和有益效果:
(1)本发明调控了二氧化铈催化剂的氧空位浓度,提高其氧化还原性。
(2)本发明调控了二氧化铈催化剂的表面酸量与酸强度,提高其抗积碳能力。
(3)本发明的三维有序大孔氧缺陷型二氧化铈催化剂表现出良好光热催化活性与稳定性。
(4)本发明的原料廉价易得,制备过程简单,成本低。
附图说明
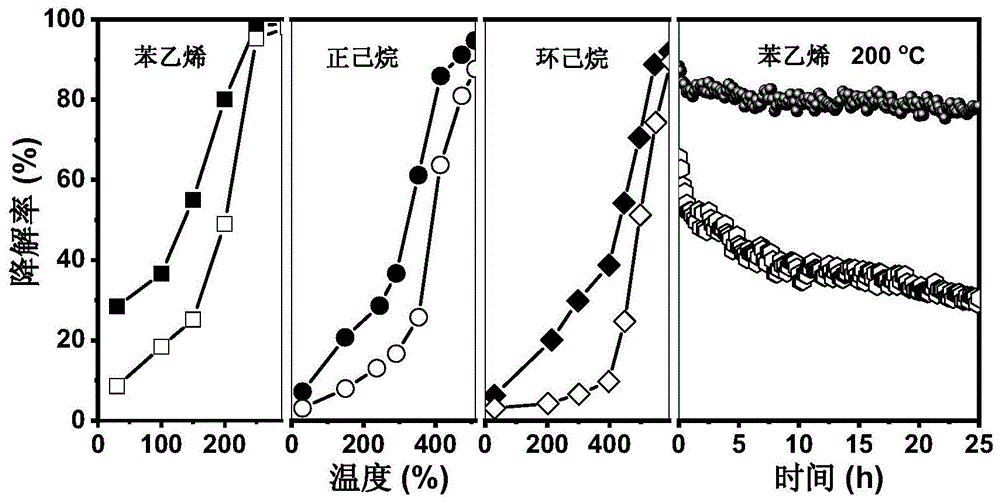
图1为三维有序大孔氧缺陷型二氧化铈催化剂对气相苯乙烯、正己烷和环己烷的光热催化降解曲线和稳定性测试图,其中实心标志图为三维有序大孔氧缺陷型二氧化铈催化剂1,空心标志图为三维有序大孔二氧化铈催化剂1。
具体实施方式
以下通过具体实施方式的描述对本发明作进一步说明,但这并非是对本发明的限制,本领域技术人员根据本发明的基本思想,可以作出各种修改或改进,但是只要不脱离本发明的基本思想,均在本发明的范围之内。若未特别指明,实施例中所用的技术手段为本领域技术人员所熟知的常规手段。除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备。
实施例1
s1、取等摩尔(2mmol)的六水合硝酸铈与一水柠檬酸粉末装入玻璃烧杯中,加入1.2ml醇溶液(乙二醇和甲醇体积比为1:1),常温超声1h后得到均匀透明溶液a。
s2、将0.5g的pmma微球粉末(直径为300nm)浸渍在溶液a中,超声混合5min,避光静置12h后,用砂芯过滤装置进行真空抽滤,以除去多余液体,得到固体b,并对固体b进行60℃真空干燥12h,然后将其移入管式炉,在氩气氛围下130℃煅烧1h后,以1℃/min的速率升温至600℃,并在600℃煅烧5h,随后在空气中600℃煅烧3h,得到固体c。
s3、将所得到的固体c移入固定床反应器,采用氮气鼓泡装置控制反应器内湿度至30%,在氢气-氮气混合气(氢气与氮气体积比为1:1)氛围下400℃煅烧2h;然后在保持400℃条件下通入氮气吹扫1h进行除湿处理,随后在空气-氮气混合气(空气与氮气体积比为1:1)氛围下400℃煅烧2h,得到三维有序大孔氧缺陷型二氧化铈催化剂1。
实施例2
s1、取2mmol六水合硝酸铈与8mmol一水柠檬酸粉末装入玻璃烧杯中,加入2.5ml醇溶液(乙二醇和甲醇体积比为1:5),常温超声0.5h后得到均匀透明溶液a。
s2、将1g的pmma微球粉末(直径为200nm)浸渍在溶液a中,超声混合5min,避光静置24h后,用砂芯过滤装置进行真空抽滤,以除去多余液体,得到固体b,并对固体b进行30℃真空干燥96h,然后将其移入管式炉,在氩气氛围下100℃煅烧3h后,以5℃/min的速率升温至400℃,并在400℃煅烧36h,随后在空气中400℃煅烧36h,得到固体c。
s3、将所得到的固体c移入固定床反应器,采用氮气鼓泡装置控制反应器内湿度至80%,在氢气-氮气混合气(氢气与氮气体积比为1:8)氛围下400℃煅烧10h;然后在保持400℃条件下通入氮气吹扫1h进行除湿处理,随后在空气-氮气混合气(空气与氮气体积比为1:8)氛围下400℃煅烧10h,得到三维有序大孔氧缺陷型二氧化铈催化剂2。
实施例3
s1、取2mmol六水合硝酸铈与0.5mmol一水柠檬酸粉末装入玻璃烧杯中,加入1ml乙醇溶液,常温超声0.5h后得到均匀透明溶液a。
s2、将0.4g的pmma微球粉末(直径为500nm)浸渍在溶液a中,超声混合5min,避光静置8h后,用砂芯过滤装置进行真空抽滤,以除去多余液体,得到固体b,并对固体b进行80℃真空干燥1h,然后将其移入管式炉,在氩气氛围下130℃煅烧1h后,以5℃/min的速率升温至800℃,并在800℃煅烧2h,随后在空气中800℃煅烧6h,得到固体c。
s3、将所得到的固体c移入固定床反应器,采用氮气鼓泡装置控制反应器内湿度至20%,在氢气-氮气混合气(氢气与氮气体积比为1:0.5)氛围下600℃煅烧1h;然后在保持600℃条件下通入氮气吹扫1h进行除湿处理,随后在空气-氮气混合气(空气与氮气体积比为1:0.5)氛围下600℃煅烧1h,得到三维有序大孔氧缺陷型二氧化铈催化剂3。
实施例4
s1、取2mmol六水合硝酸铈与16mmol一水柠檬酸粉末装入玻璃烧杯中,加入4ml醇溶液(乙二醇和甲醇体积比为1:0.5),常温超声2h后得到均匀透明溶液a。
s2、将1.5g的pmma微球粉末(直径为400nm)浸渍在溶液a中,超声混合5min,避光静置24h后,用砂芯过滤装置进行真空抽滤,以除去多余液体,得到固体b,并对固体b进行60℃真空干燥36h,然后将其移入管式炉,在氩气氛围下150℃煅烧1h后,以1℃/min的速率升温至900℃,并在900℃煅烧1h,随后在空气中900℃煅烧1h,得到固体c。
s3、将所得到的固体c移入固定床反应器,采用氮气鼓泡装置控制反应器内湿度至60%,在氢气-氮气混合气(氢气与氮气体积比为1:5)氛围下600℃煅烧5h;然后在保持600℃条件下通入氮气吹扫1h进行除湿处理,随后在空气-氮气混合气(空气与氮气体积比为1:5)氛围下600℃煅烧5h,得到三维有序大孔氧缺陷型二氧化铈催化剂4。
实施例5
s1、取2mmol六水合硝酸铈与1mmol一水柠檬酸粉末装入玻璃烧杯中,加入1ml醇溶液(乙二醇和甲醇体积比为1:0.8),常温超声1h后得到均匀透明溶液a。
s2、将0.4g的pmma微球粉末(直径为50nm)浸渍在溶液a中,超声混合5min,避光静置24h后,用砂芯过滤装置进行真空抽滤,以除去多余液体,得到固体b,并对固体b进行70℃真空干燥6h,然后将其移入管式炉,在氩气氛围下150℃煅烧2h后,以1℃/min的速率升温至500℃,并在500℃煅烧24h,随后在空气中500℃煅烧36h,得到固体c。
s3、将所得到的固体c移入固定床反应器,采用氮气鼓泡装置控制反应器内湿度至50%,在氢气-氮气混合气(氢气与氮气体积比为1:0.1)氛围下200℃煅烧24h;然后在保持200℃条件下通入氮气吹扫1h进行除湿处理,随后在空气-氮气混合气(空气与氮气体积比为1:0.1)氛围下200℃煅烧24h,得到三维有序大孔氧缺陷型二氧化铈催化剂5。
对比例1
s1、取等摩尔(2mmol)的六水合硝酸铈与一水柠檬酸粉末装入玻璃烧杯中,加入1.2ml醇溶液(乙二醇和甲醇体积比为1:1),常温超声1h后得到均匀透明溶液a。
s2、将0.5g的pmma微球粉末(直径为300nm)浸渍在溶液a中,超声混合5min,避光静置12h后,用砂芯过滤装置进行真空抽滤,以除去多余液体,得到固体b,并对固体b进行60℃真空干燥12h,然后将其移入管式炉,在氩气氛围下130℃煅烧1h后,以1℃/min的速率升温至600℃,并在600℃煅烧5h,随后在空气中600℃煅烧3h,得到固体c。
s3、将所得到的固体c移入固定床反应器,在氮气氛围下200℃煅烧3h,随后在空气-氮气混合气(空气与氮气体积比为1:1)氛围下400℃煅烧2h,得到三维有序大孔二氧化铈催化剂1。
试验例1
本发明制备得到的三维有序大孔氧缺陷型二氧化铈催化剂对气相苯乙烯、正己烷和环己烷的光热催化降解曲线和稳定性测试图
实验方法:使用光热固定床反应器对催化剂的光热催化性能进行表征(该反应器一侧带有视窗,能将光线照射进去)。①活性测试方法:取100mg催化剂装填到内径为6mm的石英反应管中,利用热电偶进行控温,在30~600℃间进行实验。用n2鼓泡装置产生苯乙烯、正己烷和环己烷蒸汽,并用干燥空气稀释至50ppm后通入反应管。气体总流量为50mlmin-1,空速为30,000mlh-1g-1。待催化剂在常温无光下吸附24h达到吸附-脱附平衡后,开启300w氙灯(λ=300~780nm,光强为200mw·cm-2),升温进行光热催化反应。反应尾气通入气相色谱(gc9800,fid)中进行在线分析vocs浓度。在实验过程中,每个温度取三个数据点,并且每个温度点的取样时间间隔至少在15min以上,即床层温度达到稳定,以便每次测量数据误差保持在10%以内。②稳定性测试方法:待催化剂在常温无光下吸附24h达到吸附-脱附平衡后,开启300w氙灯(λ=300~780nm,光强为200mw·cm-2),并将光热固定床反应器升温至200℃后,采用气相色谱(gc9800,fid)在线测定苯乙烯浓度,每隔10min进行采样分析。
实验结果:图1为三维有序大孔氧缺陷型二氧化铈催化剂对气相苯乙烯、正己烷和环己烷的光热催化降解曲线和稳定性测试图。该催化剂由实施例1方法制得,并将其催化性能与对比例1的三维有序大孔二氧化铈催化剂1作比。其中,经tem与xrd表征发现两者的形貌结构一致,raman与epr证实了实施例1的催化剂富含氧空位,并且与对比例1的催化剂作比,实施例1的催化剂表面酸量增多但酸强度下降(nh3-tpd表征)。由图1可以看出,与对比例1的催化剂作比,实施例1的催化剂表现出更好的光热催化活性与稳定性,降解结果表明在光照下(λ=300~780nm,光强为200mw·cm-2),苯乙烯、正己烷和环己烷完全降解(>95%)的温度分别为250℃、510℃和590℃。在实施例1的催化剂200℃光热条件下反应25h内,苯乙烯的降解率保持在75%以上,而对比例1的催化剂在反应25h后其催化性能下降了50%。说明在二氧化铈催化剂的三维有序大孔结构、氧空位与酸性位的协同作用下,能有效增强催化剂的氧化还原性与稳定性。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!