一种超低本底金特效树脂及其制备和应用的制作方法
本发明属于地球化学领域,具体涉及一种超低本底金特效树脂及其制备和应用。
背景技术:
贵金属,比如金、钯、铂,尤其是金,其在行星演化,壳幔分异等重大地质问题研究中也发挥着非常重要的作用,因此受到地球科学家的广泛关注。但是,金在地壳中的丰度极低,且分布不均,这给金的分离、富集和分析带来了很大的困难。
目前,常用的金的富集分离方法有共沉淀分离、活性炭吸附富集分离法、溶剂萃取富集分离法、泡沫塑料富集分离法、载体泡塑富集分离法及离子交换树脂富集分离法等几种。但是采用共沉淀分离,其本底非常高,流程也非常复杂,不适合低含量样品的分析。液液萃取,由于采用大量的萃取剂,污染高,同时大量萃取的引入造成流程空白高,并有一定的样品损失。聚氨酯对金有非常好的选择性,广泛应用于对金的分析。然而,聚氨酯泡塑是一种多孔性高分子材料,分析工作者应用泡塑分离富集金的时候,一般选用静态分离方式,即:将聚氨酯泡塑投入样品的消解液中,在往复振荡机上振荡一定时间,将聚氨酯泡塑从消解液中捞出,用水冲洗掉聚氨酯泡塑上粘附的样品残渣后,将其直接投入近沸的盐酸和硫脲混合液中,解脱聚氨酯泡塑上吸附的金。从这一流程中看出,整个分析过程较为粗放。对于低含量样品,采用静态振荡的方式,不能保证全部回收金,采用静态解脱的方式也不能保证金解脱的回收率。对于金而言,金只有一个同位素,必须保证高的回收率,才能获得准确的分析结果。离子交换树脂方法一般分为两种:阴、阳离子交换树脂。阳离子交换树脂以jarvis发展的方法应用最为广泛,但是树脂用量很大,如果样品中fe、ni、cu等基体元素含量较高,则需加大树脂用量,并且时常不能完全分离gd、tb、dy、ta、hf、hg等干扰元素,甚至需要再过一次阴离子柱,同时采用阳离子树脂分离au拖尾严重,耗酸耗时;阴离子树脂用量较少,但是金非常难于淋洗,淋洗金需要消耗大量的酸。
载体泡塑富集分离法是一种利用负载有螯合剂或萃取剂的泡塑对金进行富集分离的方法,兼有萃取和泡塑吸附两种功能,因而对金会有更大的富集能力。载体泡塑富集分离法有效避免了活性炭吸附富集分离法的缺点。如:活性炭解析时间长、解析率低及能耗和生产成本高的问题。溶剂萃取富集分离法的有臭味、环境污染的问题,以及离子交换树脂富集分离方法的选择性差、不易解脱的问题等。泡塑分离方法已成为金的富集分离广泛应用的新工艺方法。如专利cn201510551056.0公开了一种地球化学样品中痕量金、银、钯同时测定的方法,该专利为一种负载二苯硫脲泡塑富集分离测定金的方法,二苯硫脲以物理附着的形式负载到泡塑上,可以有效富集目标元素。但是该专利方法在制备负载二苯硫脲聚氨酯泡塑前期进行了预处理,因二苯硫脲与泡塑间的物理吸附作用比较弱,在多次振荡吸附时,仍不可避免地存在着脱落现象,且二苯硫脲与金形成的配合物结构不稳定,这会使测试结果不稳定、精密度降低,偏差很大。同时也有科研工作者研究制备高分子本身就具有一定的选择性和吸附性的树脂,如专利cn201610982022.1公开了一种改进的5-胺甲基尿嘧啶修饰泡塑富集金的分析方法,该技术采用5-胺甲基尿嘧啶做为修饰配体,以二醛为连接臂,在水相中合成5-胺甲基尿嘧啶修饰泡塑。5-胺甲基尿嘧啶和氨基泡塑分别和二醛生产亚胺,将5-胺甲基尿嘧啶以共价键的形式修饰到泡塑表面。这种方法一定程度上降低了测试偏差,相应的提高了准确度和精密度,但修饰配体与金形成的配合物结构不稳定,不能够精确地测量样品中的超痕量金。akbuluth等人(akbuluth,yamadas,endot.preparationofazwitterionicpolymerbasedonl-cysteineforrecoveryapplicationofpreciousmetals[j].rscadvances,2016,6(110).)以l-半胱氨酸和4-乙烯基氯化苄为原料,在水溶液中合成了l-半胱氨酸接枝聚苯乙烯,并将其作为水介质中钯(ⅱ)、铂(ⅳ)、金(ⅲ)离子的回收材料;dingshuaixue等人(dingshuaixue,tingli,yanhongliu,yuehengyang,yuxingzhang,juncui,dongbenguo.selectiveadsorptionandrecoveryofpreciousmetalionsfromwaterandmetallurgicalslagbypolymerbrushgraphene-polyurethanecomposite[j].reactiveandfunctionalpolymers,2019,136:138-152.)以puf为基材合成了一系列聚氨酯泡沫(puf)复合材料,并发现这种树脂对au(ⅲ)等贵金属有一定的选择性和吸附性能。上述文献也都制备出了具有吸附性和选择性的高分子聚合物,但由于聚合物主链为非极性的c-c结构,在极性条件下他们对配位元素的包埋干扰作用极大地降低了配位键的形成速度和配位元素的利用率,也提高了制备成本。另外,聚氨酯泡沫材料是一种块体结构,通常用来做静态吸附。如果想进行柱色谱分离,需要将聚氨酯泡沫破碎后装填到固相萃取柱中。但是聚氨酯泡沫破碎后有很强的静电斥力,难于装柱,限制了其使用范围。
因此,综上迫切需要研发一种对金具有较高的选择性,且精密度、准确度均较高的适合超痕量金的载体泡塑快速富集分离方法。
技术实现要素:
为解决现有载体泡塑富集分离金技术存在的对金的选择性、精密度及准确度低,不适合超痕量金快速富集分离的问题,本发明将fmoc-s-三苯甲基-l-半胱氨酸(cas:103213-32-7)负载于载体树脂,得到了一种特效树脂,该特效树脂对金具有极好的选择性。另外,由于制备步骤只有一步,因此该特效树脂的本底极低,能够对超痕量的金进行地快速富集分离,并进行高精密度和准确度的分析。
本发明的第一个目的是提供一种超低本底金特效树脂,所述金特效树脂包括载体树脂和螯合剂,所述螯合剂为fmoc-s-三苯甲基-l-半胱氨酸,结构式如下所示:
半胱氨酸具有多个能与贵金属配位的杂原子,比如氧、氮和硫原子,易于与具有d空轨道的贵金属配位。根据我们的研究,在配体中加入硫原子,可以有效改善配体对金元素的吸附能力。但是天然产物l-半胱氨酸是水溶性的,不能直接用于萃取。我们通过大量的实验,预料不到地发现常用于生物领域中合成多肽的fmoc-s-三苯甲基-l-半胱氨酸(cas#:103213-32-7)用于特效树脂中的螯合剂,可以特异性和金结合,对金选择性吸附性能好,能够有效富集样品中的待检的金元素,极大提高了金的检测精度,并且检测限极低,可以进行ng/ml级别的检测,即ppb级别的检测。这一化合物极性非常低,不溶于水,而溶于丙酮、氯仿等低沸点的有机溶剂。另外,该化合物具有较多的苯环,可以与载体树脂上的苯环发生π-π共轭作用,这一作用非常有利于配体化合物牢固的吸附在载体树脂上。
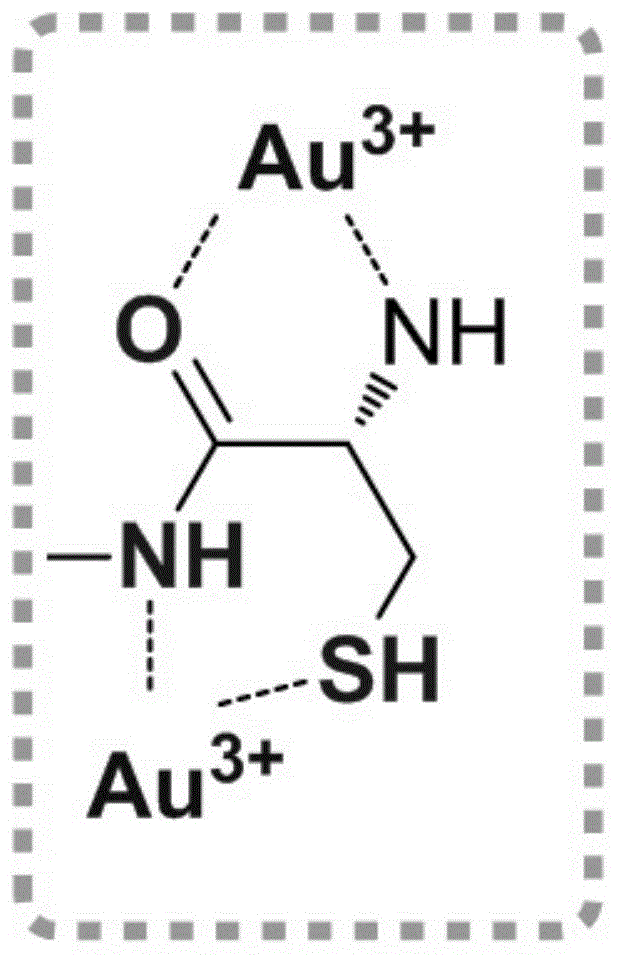
根据我们的研究,fmoc-s-三苯甲基-l-半胱氨酸易于与金配位络合并形成如图1所示的稳定的五元环构像,fmoc-s-三苯甲基-l-半胱氨酸含有大量的苯环,显著增强配体与聚合物载体直接的吸附作用,在严苛的使用条件下,配体也不容易流失,可以反复多次使用。
所述载体树脂为苯乙烯-二乙烯基苯共聚树脂或甲基丙烯酸酯交联树脂,具体型号没有特别的限定,具体苯乙烯-二乙烯基苯共聚树脂选自cg161m、cg161c、cg300s、cg300m、cg300c,甲基丙烯酸酯交联树脂选自cg71s、cg71m和cg71c。
优选地,所述载体树脂和螯合剂的体积质量比(ml/g)为10-20:1-2。
本发明的第二个目的在于提供了所述超低本底金特效树脂的制备方法,包括如下步骤:
s1)将螯合剂加至有机溶剂中,搅拌溶解,再加入载体树脂混合均匀,得混合物;
s2)将混合物进行热处理使溶剂完全挥发,得特效树脂,所得特效树脂密封在醇溶液中,并保存于聚烯烃罐中。
优选地,步骤s1)中所述有机溶剂为丙酮或氯仿中的至少一种,所述载体树脂和螯合剂的体积质量比(ml/g)为10-20:1-2。
步骤s2)所述热处理方式包括在红外灯下搅拌或者真空烘干;所述红外灯的功率为1000-2000w;所述真空干燥的温度为40-60℃,真空度为0.01-0.1mpa。
所得特效树脂密封在醇溶液中,并保存于聚烯烃罐中。所述醇优选为乙醇、丙醇、丁醇中的至少一种,所述聚烯烃优选为聚乙烯、聚丙烯。
本发明还提供了另一种制备金特效树脂的制备方法,包括如下步骤:
l1)将螯合剂加至有机溶剂中,搅拌溶解,再将其逐滴加入到载体树脂中,保存搅拌速度恒定,混合均匀,得混合物;
l2)将浓盐酸滴加至步骤l1)所得混合中,并不断搅拌,得到所述金特效树脂,直接装填入固相萃取柱中。
步骤l1)中所述有机溶剂为丙酮或氯仿中的至少一种,溶剂的用量没有特别的限定,能够将螯合剂充分分散即可,一般有机溶剂和螯合剂的体积质量比(ml/g)为10-20:1-2。
所述载体树脂和螯合剂的体积质量比(ml/g)为10-20:1-2。
所述浓盐酸的摩尔浓度为5-6mol/l,浓盐酸和螯合剂的体积重量比(ml/g)为50-100:1-5。
本发明第三个目的在于所述金特效树脂在检测样品金含量的方法,其特征在于,使用上述金特效树脂对样品中金进行富集分离后,进行金的检测。
首先需要进行淋洗条件的筛选,包括以下步骤:
n1)树脂装柱:将金特效树脂装填入固相萃取柱中,得萃取柱;
n2)预洗:用浓盐酸和超纯水依次对步骤n1)所得萃取柱进行预洗,再用稀盐酸1进行柱平衡;
n3)淋洗曲线的确定:将待测样品溶液用稀盐酸1稀释,作为上柱液,上样后分别用稀盐酸2和稀盐酸3继续过柱,最后用浓盐酸洗脱,洗脱液最终采用电感耦合等离子质谱进行检测;
所述固相萃取柱的高径比为10-15:1,优选为10-12:1。
预先配置好的待测样品溶液用稀盐酸稀释后,为含有基体元素fe、ca、mg、al(其中fe的浓度为10μg·ml-1,ca、mg、al的浓度为1μg·ml-1)和干扰元素gd、tb、dy、ta、hf、hg(浓度为100ng·ml-1)的含金溶液(au浓度为1ng·ml-1)
所述浓盐酸的浓度为4-7mol/l,优选为5-6mol/l,稀盐酸1-3的浓度独立为0.1-0.5mol/l,优选为0.1-0.3mol/l。
发明人经过大量实验,发现了优选的淋洗条件,在上述条件下可以实现对样品中痕量金的检测,同时基体元素和杂质影响小,不干扰分析测试,可以实现超低本底痕量金属的检测。
更为优选的条件是在n2)的步骤中,稀盐酸1浓度为0.1-0.2mol/l,所述特效树脂、浓盐酸、超纯水、稀盐酸1的体积比为1:5-8:15-20:4-6。
n3)步骤中,稀盐酸2浓度和步骤n2)中稀盐酸1浓度一致,为0.1-0.2mol/l,稀盐酸3浓度比稀盐酸2稍高,浓为0.3-0.4mol/l,所述特效树脂、稀盐酸1、稀盐酸2、稀盐酸3和浓盐酸的体积比为1:3-5:4-6:8-12:8-12。
得到优选的预洗和淋洗曲线条件后,以相同的条件,利用本发明得到的超低本底金特效树脂对样品中金进行富集分离后,进行检测。
所述金特效树脂在检测样品金含量的方法包括以下步骤:将样品进行消解后,用稀盐酸定容,取上清液加入包含所述金特效树脂的固相萃取柱中,进行金的富集和分离,最后将洗脱液在电感耦合等离子质谱上测定。
更为具体地,所述金特效树脂在检测样品金含量的方法包括以下步骤:
s1)用浓盐酸和超纯水依次对固相萃取柱进行预洗,再用稀盐酸1进行柱平衡;
s2)将消解的待分离纯化的的样品用稀盐酸1定容后作为上柱液,上样后分别用稀盐酸2和稀盐酸3继续过柱,最后用浓盐酸洗脱,洗脱液最终采用电感耦合等离子质谱进行检测。
所述浓盐酸的浓度为4-7mol/l,优选为5-6mol/l,稀盐酸1-3的浓度独立为0.1-0.5mol/l,优选为0.1-0.3mol/l。
优选地,所述s1)的预洗步骤中,稀盐酸1浓度0.1-0.2mol/l,所述特效树脂、浓盐酸、超纯水、稀盐酸的体积比为1:4-6:15-20:5-8;
进一步优选地,所述s2)淋洗曲线确定步骤中,稀盐酸2浓度和预洗步骤中稀盐酸1浓度一致,为0.1-0.2mol/l,稀盐酸3浓度比稀盐酸2稍高,浓度为0.3-0.4mol/l,所述特效树脂、稀盐酸1、稀盐酸2、稀盐酸3和浓盐酸的体积比为1:3-5:4-6:8-12:8-12。
所述样品的消解是本领域技所熟知的,根据不同样品的特点,可以采用不同的方法处理,比如含有金钯铂的样品,使用王水进行分解;含有粗铜的样品,先用稀硝酸处理再用王水进行分解。具体而言,本发明采用的消解方法包括以下步骤:将含有待测样品的坩埚置于马弗炉中,650~700℃灼烧1小时,取出冷却,将样品倒入带螺纹盖的特氟龙烧杯中,加少量水润湿,再加入50wt%王水和hf,打开盖,置于电热板上加热至近干,所得样品置于电热板上重复加入少量50wt%王水进一步溶解不容物,蒸至近干,最后用稀盐酸定容,定容后待测样品浓度为30-50mg/ml。
与现有技术相比,本发明的有益效果是:
一、本发明创造性地利用生物领域常用的一种中间体fmoc-s-三苯甲基-l-半胱氨酸与载体树脂复合,得到了一种能够对样品中痕量金进行有效分离富集的特效树脂,对金选择性吸附,样品中基体元素和干扰元素不会产生干扰,采用该特效树脂可以极大提高了分析的精密度和准确度,回收率均在99%左右。
二、本发明特效树脂制备工艺简单,通过螯合剂和载体树脂中搅拌即可一步完成,生产成本低。
三、在对样品进行金的富集分离测试时,该特效树脂用量较少,因此全流程空白极低,可以对超痕量样品中的金进行检测,检测限极低,方法检测限低至1.2pgml-1(3倍的7次全流程空白的标准偏差),方法定量限低至3.6pgml-1。
附图说明
图1为本发明金特效树脂所用到螯合剂和金形成的的五元环配位构象示意图。
图2为本发明所得金特效树脂对au与基体元素和干扰元素的淋洗曲线。
具体实施方式
下面结合具体实施例对本发明作进一步的说明,但并不局限于说明书上的内容。若无特殊说明,本发明实施例中所述“份”均为重量份。所用试剂均为本领域可商购的试剂。
本发明所用螯合剂fmoc-s-三苯甲基-l-半胱氨酸来自于商购,具体采购自sigma-aldrich公司。
实验用试剂和器皿的准备:样品前处理工作都需要在超净实验室中完成。实验中所用的盐酸,硝酸和氢氟酸需要通过savillex亚沸蒸馏器进行二次纯化;实验中所用蒸馏水需要经过millipore超纯水系统进行二次纯化,制备出二次超纯水。实验中所用的瓷坩埚需要用二次水浸泡过夜,用清洗过的塑料夹夹出后,再用二次超纯水多次清洗内部,并在烘箱中烘干。实验中所用的特氟龙烧杯(聚四氟乙烯)和固相萃取柱(聚丙烯塑料),在使用前需要按照下列流程严格清洗。将特氟龙烧杯(聚四氟乙烯)和固相萃取柱(聚丙烯塑料)浸泡如30%的硝酸溶液(硝酸溶液采用二次纯化的硝酸和二次超纯水配制)中过夜。然后用清洗过的塑料夹夹出后,用二次超纯水反复清洗,再用二次超纯水浸泡过夜。采用二次超纯水清洗和浸泡需要反复三次。然后,烘干备用。
本发明实施例所用固相萃取柱采购自日本室町公司,具体规格是内径5mm,长度50mm,即高径比10:1。
制备例超低本底特效树脂的制备
制备例1
超低本底金特效树脂的制备方法,包括如下步骤:
s1)将50g螯合剂fmoc-s-三苯甲基-l-半胱氨酸加至500ml丙酮中,搅拌溶解,再加入500ml苯乙烯-二乙烯基苯共聚树脂cg161c混合均匀,得混合物;
s2)将混合物进行真空干燥,温度为60℃,真空度为0.08mpa,使溶剂完全挥发,所得特效树脂密封在3.0%的乙醇溶液中,并保存于聚丙烯罐中。
制备例2
超低本底金特效树脂的制备方法,包括如下步骤:
s1)将50g螯合剂fmoc-s-三苯甲基-l-半胱氨酸加至1000ml丙酮中,搅拌溶解,再加入700ml苯乙烯-二乙烯基苯共聚树脂cg300s混合均匀,得混合物;
s2)将混合物在红外灯下1000~2000w,边加热边搅拌,使溶剂完全挥发,得特效树脂,所得特效树脂密封在3.0%乙醇中,并保存于聚丙烯罐中。
制备例3
超低本底金特效树脂的制备方法,包括如下步骤:
s1)将50g份螯合剂fmoc-s-三苯甲基-l-半胱氨酸加至500ml氯仿中,搅拌溶解,再加入500ml苯乙烯-二乙烯基苯共聚树脂cg161c混合均匀,得混合物;
s2)将100ml6mol/l的hcl逐渐滴加在上述混合物中,不断搅拌,所得特效树脂可直接装入固相萃取柱中。
制备例4
超低本底金特效树脂的制备方法,包括如下步骤:
s1)将50g份螯合剂fmoc-s-三苯甲基-l-半胱氨酸加至1000ml氯仿中,搅拌溶解,再加入1000ml苯乙烯-二乙烯基苯共聚树脂cg161c,混合均匀,得混合物;
ss2)将混合物进行真空干燥,温度为60℃,真空度为0.08mpa,使溶剂完全挥发,得特效树脂,所得特效树脂密封在在1%乙醇溶液中,并于聚丙烯罐中进行保存。
实施例样品中金的富集分离和测试
实施例1金特效树脂对au以及其它元素的淋洗曲线
将制备例1得到的1ml金特效树脂装填入固相萃取柱,先用4ml6moll-1hcl和20ml超纯水预洗该树脂,再用6ml0.1moll-1hcl溶液进行柱平衡。预先配置好的含有基体元素和干扰元素的含金溶液用盐酸调节其酸度为0.1moll-1,配制好的溶液中各元素的浓度大致为:fe的浓度为10μg·ml-1,ca、mg、al的浓度为1μg·ml-1,干扰元素gd、tb、dy、ta、hf、hg浓度为100ng·ml-1,au浓度为1ng·ml-1。取3ml样品上样过柱,先用4ml0.1moll-1稀盐酸淋洗和8ml0.3moll-1稀盐酸淋洗,最后,用8ml6moll-1hcl将金洗脱全部回收。实施例1的淋洗曲线如图2所示。通过图2可以看到,基体元素和干扰元素上述淋洗条件下,从金特效树脂中洗脱出来,而金基本留在特效树脂上,完成对金的选择性吸附,从而可以有效完成对样品中金含量的准确测试。
实施例2
利用制备例1的特效树脂对超低含量样品中的金进行分离富集。
按照实施例1相同的条件和操作对特效树脂进行处理,将制备例1得到的1ml金特效树脂装填入固相萃取柱,先用4ml6moll-1hcl和20ml超纯水预洗该树脂,再用6ml0.1moll-1hcl溶液进行柱平衡。
准确称取5.0g国际标准物质wgb-1于瓷坩埚中,将瓷坩埚置于低温马弗炉中,升温到650-700℃灼烧1小时,取出冷却,将样品倒入200ml带螺纹盖的特氟龙烧杯中,加10ml二次超纯水润湿,再加入40ml50%王水,和5mlhf,搅匀,打开盖,置于电热板上加热溶解至近干,反复2次。用0.1moll-1hcl稀盐酸定容至100ml,取3ml上清液,上柱于1ml制备例1得到的金特效树脂,按照实施例1中的淋洗曲线相同的条件进行淋洗分离,即用4ml0.1moll-1稀盐酸、8ml0.3moll-1稀盐酸、8ml6moll-1hcl依次淋洗,最后收集8ml6moll-1hcl的淋洗液,将金全部回收将洗脱液在电感耦合等离子质谱上测定。
选取国际标准物质wgb-1对上述测定方法进行可行性评价,测量次数为5次。
利用上述测定方法对国际标准物质wgb-1中金的测定值与标准值的对比如表1所示:
表1国际标准物质wgb-1中金的测定值与标准推荐值的对比
测定值以测定平均值±标准偏差的形式表达,在测定值和标准推荐值接近的情况下,标准偏差越低,说明检测的精密度越高。从表1中的实验数据可知,本发明特效树脂采用了螯合剂fmoc-s-三苯甲基-l-半胱氨酸,对分析方法的准确度没有影响,极大提高了检测的精密度,回收率高达99.2%,说明本发明以一种简单的方式即可满足科研工作的要求,不需要复杂的设备和成本高昂的试剂,具有优异的可靠性和产业上的可行性。
实施例3
按照实施例1相同的条件和操作对特效树脂进行处理,区别在于待测样品为5.0g国际标准物质tdb-1。
选取国际标准物质tdb-1对上述测定方法进行可行性评价,测量次数为5次。利用上述测定方法对国际标准物质tdb-1中金的测定值与标准值的对比如表2所示:
表2国际标准物质tdb-1中金的测定值与标准推荐值的对比
实施例4
按照实施例1相同的条件和操作对特效树脂进行处理,区别在于待测样品为5.0克国际标准物质wpr-1。
选取国际标准物质wpr-1对上述测定方法进行可行性评价,测量次数为5次。利用上述测定方法对国际标准物质wpr-1中金的测定值与标准值的对比如表3所示:
表3国际标准物质wpr-1中金的测定值与标准推荐值的对比
实施例5
其他同实施例2相同,不同之处在于所用金特效树脂为制备例2得到的。
按照实施例1相同的条件和操作对特效树脂进行处理。之后选取国际标准物质wgb-1对上述测定方法进行可行性评价,测量次数为5次。利用上述测定方法对国际标准物质wgb-1中金的测定值与标准值的对比如表4所示:
表4国际标准物质wgb-1中金的测定值与标准推荐值的对比
实施例6
其他同实施例2相同,不同之处在于所用金特效树脂为制备例3得到的。
按照实施例1相同的条件和操作对特效树脂进行处理。之后选取国际标准物质wgb-1对上述测定方法进行可行性评价,测量次数为5次。利用上述测定方法对国际标准物质wgb-1中金的测定值与标准值的对比如表5所示:
表5国际标准物质wgb-1中金的测定值与标准推荐值的对比
实施例7
其他同实施例2相同,不同之处在于所用金特效树脂为制备例4得到的。
按照实施例1相同的条件和操作对特效树脂进行处理。之后选取国际标准物质wgb-1对上述测定方法进行可行性评价,测量次数为5次。利用上述测定方法对国际标准物质wgb-1中金的测定值与标准值的对比如表6所示:
表6国际标准物质wgb-1中金的测定值与标准推荐值的对比
实施例8
其他同实施例2相同,不同之处在于淋洗条件改为:将制备例1得到的金特效树脂装填固相萃取柱(对应的柱床体积为1ml),先用4ml6moll-1hcl和超纯水预洗该树脂,再用6ml0.1moll-1hcl溶液进行柱平衡,再用12ml0.1moll-1稀盐酸淋洗,最后用8ml6moll-1hcl将金全部回收。
选取国际标准物质wgb-1对上述测定方法进行可行性评价,测量次数为5次。利用上述测定方法对国际标准物质wgb-1中金的测定值与标准值的对比如表7所示:
表7国际标准物质wgb-1中金的测定值与标准推荐值的对比
实施例9
其他同实施例2相同,不同之处在于淋洗条件改为:将制备例1得到的金特效树脂装填固相萃取柱(对应的柱床体积为1ml),先用4ml6moll-1hcl和超纯水预洗该树脂,再用6ml0.1moll-1hcl溶液进行柱平衡,再用12ml0.2moll-1稀盐酸淋洗,最后用8ml6moll-1hcl将金全部回收。
选取国际标准物质wgb-1对上述测定方法进行可行性评价,测量次数为5次。
利用上述测定方法对国际标准物质wgb-1中金的测定值与标准值的对比如表9所示:
表9国际标准物质wgb-1中金的测定值与标准推荐值的对比
实施例10
萃取柱的准备:将制备例1得到的金特效树脂1ml装填入固相萃取柱,用4ml浓盐酸(6mol/l)和8ml超纯水依次对固相萃取柱进行预洗,再用8ml稀盐酸(0.1mol/l)进行柱平衡;
全流程空白:即不加样品的全流程收集液可以作为过程空白使用。10ml二次超纯水倒入200ml带螺纹盖的特氟龙烧杯中,再加入40ml50%王水,和5mlhf,搅匀,打开盖,置于电热板上加热溶解至近干,反复2次。用0.1moll-1hcl稀盐酸定容至100ml。再取3ml上清液,上柱。按照实施例1中的淋洗曲线相同的条件进行淋洗分离,即用4ml0.1moll-1稀盐酸、8ml0.3moll-1稀盐酸、8ml6moll-1hcl依次淋洗,最后收集8ml6moll-1hcl的淋洗液,将最后回收的8ml洗脱液在电感耦合等离子质谱上测定。
仪器条件:采用thermofisherscienticelementiicp-ms;采用micromist微量雾化器,流速0.2mlmin-1;rt功率1300w;冷却器流量15lmin-1;辅助气流量1.0lmin-1;载气流量0.90lmin-1;样品提升量200μlmin-1;取样锥1.1mm(ni);截取锥0.8mm(ni);检测模式:脉冲计数;清洗时间:60秒。
检测限:方法检测限和方法定量限根据国际纯粹与应用化学联合会(iupac)给出的方法进行计算,采用制备例1得到的特效树脂,方法检测限低至1.2pgml-1(3倍的7次全流程空白的标准偏差),方法定量限低至3.6pgml-1(10倍的7次全流程空白的标准偏差)。检测结果如下表10所示,单位均为pgml-1。
表10
上述详细说明是针对本发明其中之一可行实施例的具体说明,该实施例并非用以限制本发明的专利范围,凡未脱离本发明所为的等效实施或变更,均应包含于本发明技术方案的范围内。
- 还没有人留言评论。精彩留言会获得点赞!