一种黄酮类化合物及其结构类似物的合成方法与流程
本发明属于化学合成领域,具体涉及一种黄酮类化合物及其结构类似物的合成方法。
背景技术:
2-取代苯并-γ-吡喃酮也称2-取代黄酮,在植物中广泛存在,具有抗炎、抗癌、抗氧化、抗病毒、解痉、雌激素样作用等生物活性,在医药领域应用广泛。2-取代-4-喹诺酮是2-取代苯并-γ-吡喃酮的结构类似物,二者的差异在于杂环上的杂原子不同:前者为o,后者为n。2-取代-4-喹诺酮类化合物也广泛存在于自然界中,具有多种生物和生理活性,被广泛用作药物,如抗菌活性的代表药物诺氟沙星、抗病毒活性的代表药物为埃替格韦等。
然而,从天然产物中得到的2-取代苯并-γ-吡喃酮类化合物和2-取代-4-喹诺酮类化合物,种类有限、结构多样性少,无法完全满足医药用途。因此,化学合成上述化合物成为获得新颖结构的2-取代苯并-γ-吡喃酮和2-取代-4-喹诺酮化合物、发现新药的重要途径。
目前,文献报道构建2-取代苯并-γ-吡喃酮类化合物的方法主要有:(1)以2-羟基查耳酮为底物,酸性条件下关环,再经氧化剂如碘(i2)、二氯二氰基苯醌(ddq)等氧化得到(angew.chem.int.ed.2012,51,13070-13074;org.lett.,12(10),2010)。该方法所用原料价廉易得、反应简单,但是底物适应性差、反应中产生的副产物较多。(2)以苯乙炔、一氧化碳、乙酸2-碘苯酯为底物,经有机金属催化剂偶联成苯基丙炔酮类化合物,再在酸性条件下关环得到(tetrahedron,67(51),9993-9997;2011;j.org.chem.2008,73,1620-1623)。该法反应条件温和可在室温下进行,但是反应的前驱体不易制备,且所用金属催化剂残留高不利于分离。(3)以苯乙酮和苯甲酰卤为原料,经偶联重排成β-二酮类化合物,在酸性条件下关环得到(phytochemistry(1988),28,(1),295-6.;tetrahedronletters,57(34),3841-3843;2016)。该法有较高的产率,但是反应路线较长,会用到有腐蚀性的酸,对设备要求较高。
构建2-取代-4-喹诺酮类化合物的方法主要有:(1)以2-氨基查耳酮为原料经关环氧化得到2-取代-4-喹诺酮(bull.koreanchem.soc.2008,29(9),1853-1856.)。该方法所用原料价廉易得、反应简单,反应中产生的副产物较多。(2)亚胺中间体路径,以苯胺和β-酮酸酯加热生成亚胺,再在高温条件下关环成2-取代-4-喹诺酮类化合物(j.am.chem.soc.1939,61(10),2890-2895.;tetrahedron2018,74(52),7408-7420.)。该法原料价廉易得,但是反应需要极高的温度,条件苛刻难以操作。(3)以2-氨基(胺基)苯基丙炔酮为底物经金属催化剂关环得到2-取代-4-喹诺酮类化合物(eur.j.org.chem.2014,(19),4044-4052)。该法条件温和,但是反应的前驱体不易制备,且所用金属催化剂残留高不利于分离。(4)酰胺中间体路径,以邻氨基苯乙酮与苯甲酰卤或邻卤苯乙酮与苯甲酰胺为底物经偶联、关环得到2-取代-4-喹诺酮类化合物(tetrahedronletters58(2017)279–284)。该法反应路线长,收率低,且会用到有腐蚀性的酸酐。
从上述文献可以看出,2-取代苯并-γ-吡喃酮类化合物与2-取代-4-喹诺酮类化合物的合成,虽然起始物有差异,但形成骨架的前驱体类似,二者都可以经相应的查耳酮,经催化关环氧化得到对应的目标化合物。
1986年,日本学者yokiohoshino等利用二苯基二硫作为催化剂、2-羟基查耳酮为原料,在250~290℃下通氮气流反应2~3小时,合成了一系列2-取代并-γ-吡喃酮类化合物。该法产物单一,产率较高,但同时也存在反应温度过高、操作十分困难等缺点。而且在此高温下,大部分原料查尔酮会发生升华现象,导致其被氮气流带出反应体系、降低产物收率;与此同时,反应过程中产生的有毒且具有恶臭气味的苯硫酚也被带离出反应体系,不仅污染环境,而且对人体极为有害。这不符合绿色化学的理念,因此,此方法未能得到广泛应用。
综上,开发一种高效、环保、低廉易操作的2-取代苯并-γ-吡喃酮和2-取代4-喹诺酮类化合物合成方法,具有重要的现实意义。
技术实现要素:
本发明的目的是提供一种全新的高效、环保、低廉易操作的2-取代苯并-γ-吡喃酮和2-取代4-喹诺酮类化合物的合成方法。
为实现上述发明目的,本发明所采用的技术方案是:一种用于黄酮类化合物及其结构类似物的合成反应中的催化剂,所述催化剂为巯基功能化sba-15型介孔材料,其结构通式为:
优选的,所述巯基功能化sba-15型介孔材料中,巯基的摩尔含量为5~30%。
优选的,所述巯基功能化sba-15型介孔材料中,巯基的摩尔含量为15%。
相应的,一种黄酮类化合物及其结构类似物的合成方法,以巯基功能化sba-15型介孔材料为催化剂,以2-羟基查耳酮及其衍生物或2-氨基查耳酮及其衍生物为原料进行合成。
优选的,所述巯基功能化sba-15型介孔材料的结构通式为:
优选的,所述2-羟基查耳酮及其衍生物、2-氨基查耳酮及其衍生物的结构通式为:
x=o,n=1或x=n,n=2;
r1、r2为芳基、酰基、酰胺基、酯基、磺酸基、磺酸胺基、磺酸酯基、亚磺酸基、亚磺酸胺基、亚磺酸酯基、腈基、烷基、环烷基、烯基、炔基、卤素、烷氧基、氨基、胺基、硫醚基、硝基、磷酸酯基中的任意一种或多种。
优选的,所述合成方法包括如下步骤:
(1)以共聚法制备巯基功能化sba-15型介孔材料;
(2)以2-羟基查耳酮及其衍生物或2-氨基查尔酮及其衍生物为原料,以巯基功能化sba-15型介孔材料为催化剂,在无溶剂、大气环境条件下,加热至120~160℃,反应16~24小时,即得反应终产品。
优选的,所述合成方法还包括:
(3)将所述反应终产品经氯仿或甲醇洗出,旋蒸除去溶剂,干燥后即得;
或;
(3)将所述反应终产品经氯仿或甲醇洗出,除去溶剂、干燥、甲醇重结晶,即得。
相应的,利用权利要求4~8任意一项所述合成方法合成的化合物。
优选的,所述化合物的结构通式为:
x=o,n=0,或x=n,n=1。
本发明具有以下有益效果:
(1)本发明提供一种合成2-取代苯并-γ-吡喃酮类化合物及其衍生物、2-取代-4-喹诺酮及其衍生物的新方法。该方法高效、绿色、价格低廉、操作简单,对生产设备要求不高,终产品纯化处理简单,大部分纯度≥98%的终产品可以直接经氯仿或甲醇洗出,旋蒸除去溶剂干燥即可获得,或将洗出的粗产品除去溶剂后干燥经甲醇重结晶即可获得。
(2)本发明首次使用巯基功能化介孔材料作为催化剂进行2-取代苯并-γ-吡喃酮类化合物及其衍生物、2-取代-4-喹诺酮及其衍生物的合成反应。合成所述催化剂的原料价廉易得,合成方法简单;合成的催化剂无味、无毒,易于产物分离、无催化剂残留,可重复利用。同时,使用该催化剂进行合成反应,无需使用溶剂,氧化剂为氧气或大气。有效解决了反应苛刻、操作困难、选择性及产率不高、催化剂有毒害、反应副产物有毒害、反应过程释放有毒且难闻气体等问题。真正实现了这类化合物的绿色环保化生产。
附图说明
图1为本发明所述的sba-sh-y的反应过程示意图;
图2本发明的实施例中原料a的结构式示意图;
图3为本发明的实施例中制备的各终产物b的结构式示意图。
具体实施方式
本发明提供了一种2-取代苯并-γ-吡喃酮和2-取代-4-喹诺酮类化合物及其衍生物的合成方法。以共聚法制备的巯基功能化sba-15型介孔材料(以下简称sba-sh-y)为催化剂,以2-羟基查耳酮及其衍生物或2-氨基查尔酮及其衍生物为原料,在无溶剂、大气环境条件下,加热至120~160℃,反应16~24小时后得到所需物质。终产品可以直接经氯仿或甲醇洗出,旋蒸除去溶剂干燥后即可得;或将洗出的粗产品除去溶剂后干燥经甲醇重结晶后获得。
所述合成方法的反应方程式如下:
式中,x=o,n=1,m=0;或者,x=n,n=2,m=1。r1、r2独立地为一个或者多个芳基、酰基、酰胺基、酯基、磺酸基、磺酸胺基、磺酸酯基、亚磺酸基、亚磺酸胺基、亚磺酸酯基、腈基、烷基、环烷基、烯基、炔基、卤素、烷氧基、氨基、胺基、硫醚基、硝基、磷酸酯基。
本发明所用的2-羟基查耳酮及其衍生物或2-氨基查尔酮及其衍生物可以市购,也可以自行制备。其中,所述2-羟基查耳酮及其衍生物的制备反应式为:
r和r′独立地为一个或者多个芳基、酰基、酰胺基、酯基、磺酸基、磺酸胺基、磺酸酯基、亚磺酸基、亚磺酸胺基、亚磺酸酯基、腈基、烷基、环烷基、烯基、炔基、卤素、烷氧基、氨基、胺基、硫醚基、硝基、磷酸酯基。
以式中r和r′都选h为例,上述反应进行的具体操作如下:在单口反应瓶中加入2-羟基苯乙酮、苯甲醛、氢氧化钠和95%乙醇,放置于室温环境搅拌12h后,用1mlo/l稀盐酸淬灭,用乙酸乙酯萃取,盐水洗,减压旋转蒸发除去乙酸乙酯,干燥,乙醇重结晶即得。
所述2-氨基查尔酮及其衍生物的制备反应式为:
r和r′独立地为一个或者多个芳基、酰基、酰胺基、酯基、磺酸基、磺酸胺基、磺酸酯基、亚磺酸基、亚磺酸胺基、亚磺酸酯基、腈基、烷基、环烷基、烯基、炔基、卤素、烷氧基、氨基、胺基、硫醚基、硝基、磷酸酯基。
以r和r′都选h为例,具体操作如下:在单口反应瓶中加入2-氨基苯乙酮、苯甲醛、氢氧化钠和95%乙醇,放置于室温环境搅拌16h,用1mlo/l稀盐酸淬灭,用乙酸乙酯萃取,盐水洗,减压旋转蒸发除去乙酸乙酯后,再过200~300目硅胶柱分离,洗脱剂为pe/ea,即得。
需要说明的是,本法中所用2-羟基苯乙酮及其衍生物、2-氨基苯乙酮及其衍生物,还包括稠环的2-羟基苯乙酮及其衍生物和2-氨基苯乙酮及其衍生物;所用苯甲醛及其衍生物包括稠环的苯甲醛、脂肪基醛、芳杂环醛及其衍生物。
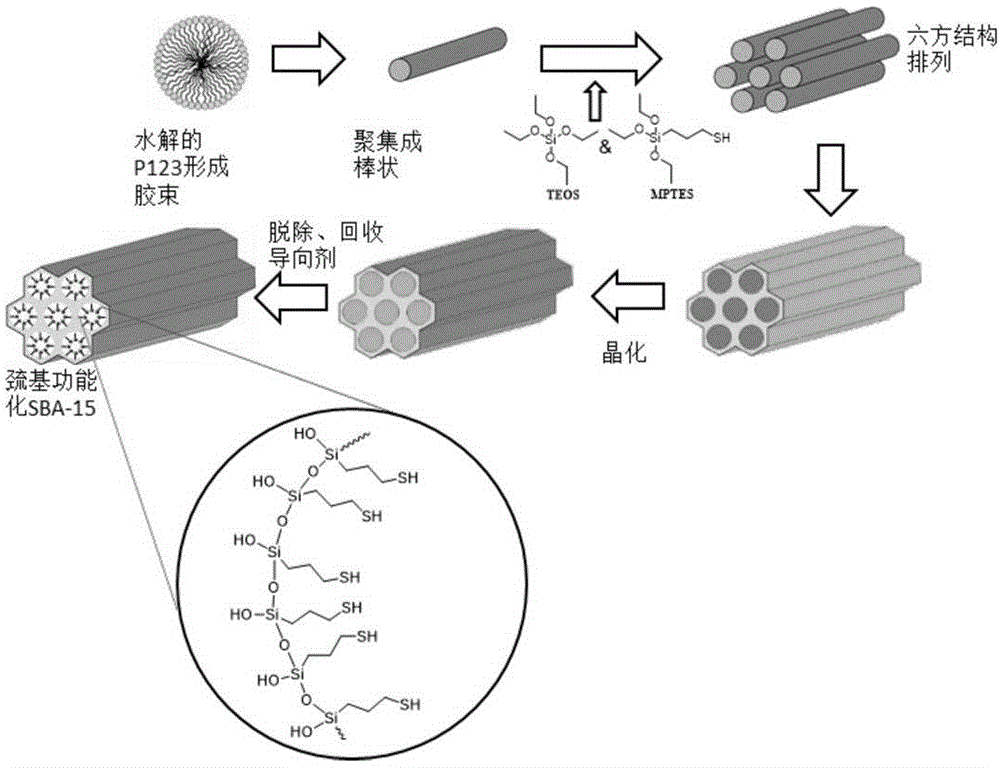
具体的,所述sba-sh-y的制备方法为:以巯基三烷氧基硅为功能前驱体,即巯基功能单体,其通式为(r3o)3-si-r4-sh,其中r3为甲基或乙基,r4为烷基或者芳基中的任意一种,优选为正丙基。结构通式如下:
在酸性条件下(ph≤1),以三嵌段非离子表面活性剂(p123)为结构导向剂,与四乙氧基硅烷(teos)或四甲基硅烷(tmos)共聚,晶化后用95%乙醇萃取回收导向剂。具体反应过程如图1所示。
优选的方案为,以共聚法制备的巯基功能化sba-15型介孔材料中,巯基的摩尔含量为5~30%,优选为15%。(以下简称巯基的摩尔含量为15%的介孔材料为sba-sh-15%)。
所述sba-sh-15%的具体制备方法为:在锥形瓶中加入3gp123,去离子水94ml和12mol/l盐酸15ml,于室温搅拌至清澈得溶液a(约3h)。将溶液a于40℃(浴温)加热1h;随后滴加teos6.15ml(27.75mmol)。滴毕,剧烈搅拌下于40℃反应45min。随后用注射器于4min内缓慢注入巯基功能单体(4.88mmol,r3为乙基,r4为正丙基),反应23h后,将反应液转移至高压反应釜,于100℃晶化30h。抽滤,滤饼用去离子水洗涤至中性后用95%乙醇(3次×10ml/次)洗涤,于室温干燥24h,得白色固体m。在圆底烧瓶中加入所述白色固体m2g和95%乙醇150ml,于60℃洗涤6h。过滤,将滤饼于室温干燥12h,用索氏提取器在95%乙醇中回流24h,除去p123,再于100℃干燥2h,得白色固体,即为巯基摩尔含量为15%的sba-sh-15%(产量1.7g)。
更优选的方案为:按质量比,所述sba-sh-15%:2-羟基查耳酮及其衍生物(或2-氨基查耳酮及其衍生物)=5~10:1。
下面结合具体实施例对本发明做进一步的详细描述。应当理解的是,这些实施例只适用于解释本发明的技术方案,并不是对发明形成任何的限制。实施例中的a3~a15的具体结构如图2所示,b3~b15如图3所示。
实施例一
在15ml单口反应管中,加入22.4mg(0.1mmol)的1-(2-羟基苯基)-3-苯基丙-2-烯-1-酮(a1),224mg的sba-sh-15%,于140℃下搅拌。约24h后,tlc检测反应完毕,加入5ml氯仿提取,抽滤,取滤液减压旋蒸除去氯仿,得到固体,将该固体用甲醇重结晶即得所需产品(b1)20mg,收率为79%。反应式如下:
b1的物理性质及谱图数据如下:白色固体。1h-nmr(400mhz,cdcl3)δ8.27(dd,j=7.9,1.5hz,1h),8.05–7.92(m,2h),7.79–7.70(m,1h),7.66–7.52(m,4h),7.51–7.42(m,1h),6.87(s,1h)。13c-nmr(101mhz,cdcl3)δ178.50,163.43,156.29,133.80,131.82,131.63,129.08,126.33,125.74,125.26,123.99,118.12,107.64。hrms(esi):m/zcalc。forc15h11o2[m+h]+:223.0759,found:223.0756。
实施例二
在15ml单口反应管中,加入25.3mg(0.1mmol)的1-(2-羟基-6-甲氧基苯基)-3-(4-甲氧基苯基)丙-2-烯-1-酮(a2),253mg的sba-sh-15%,于140℃下搅拌。约24h后tlc检测反应完毕,加入5ml甲醇提取,抽滤,取滤液减压旋蒸除去甲醇得到固体,将该固体用甲醇重结晶得产品b223mg,收率82%。反应式如下
b2物理性质及谱图数据如下:白色固体。1hnmr(400mhz,dmso-d6)δ11.60(s,1h),8.09(d,j=7.9hz,1h),7.81(d,j=8.6hz,2h),7.77(d,j=8.3hz,1h),7.66(t,j=7.6hz,1h),7.33(t,j=7.5hz,1h),7.14(d,j=8.7hz,2h),6.31(s,1h),3.86(s,3h)。13cnmr(101mhz,dmso)δ177.40,161.53,150.24,140.95,132.17,129.32,126.67,125.16,123.62,119.08,114.87,106.97,55.92。hrms(esi):m/zcalc。forc16h13no2[m+h]+:252.0980,found:252.0978。
实施例三
在15ml单口反应管中,加入25.8mg(0.1mmol)的a3,258mg的sba-sh-15%,于140℃下搅拌。约24h后tlc检测反应完毕,加入5ml氯仿提取,抽滤,取滤液减压旋蒸除去氯仿得到固体,将该固体用甲醇重结晶得产品(b3)19mg白色固体,收率76%。
b3的谱图数据如下:1hnmr(400mhz,cdcl3)δ8.26(d,j=6.9hz,1h),7.90(d,j=8.6hz,2h),7.74(dd,j=11.3,4.1hz,1h),7.60(d,j=8.4hz,1h),7.54(d,j=8.6hz,2h),7.46(t,j=7.5hz,1h),6.83(s,1h)。13cnmr(101mhz,cdcl3)δ178.30,162.25,156.19,137.91,137.86,133.94,130.27,129.41,127.57,125.78,125.41,123.93,118.06,107.72。hrms(esi):m/zcalc。forc15h10clo2[m+h]+:257.0369,found:257.0367。
实施例四
在15ml单口反应管中,加入25.4mg(0.1mmol)的a4,254mg的sba-sh-15%,于140℃下搅拌。约24h后tlc检测反应完毕,加入5ml氯仿提取,抽滤,取滤液减压旋蒸除去氯仿得到固体,将该固体用甲醇重结晶得产品(b4)20mg,收率76%,为白色固体。
b4的谱图数据如下:1hnmr(400mhz,cdcl3)δ8.25(dd,j=7.9,1.4hz,1h),7.92(d,j=8.9hz,2h),7.76–7.67(m,1h),7.58(d,j=8.3hz,1h),7.44(t,j=7.5hz,1h),7.06(d,j=8.9hz,2h),6.78(s,1h),3.92(s,3h)。13cnmr(101mhz,cdcl3)δ178.42,163.43,162.41,156.20,133.58,128.03,125.69,125.10,124.05,123.95,117.98,114.48,106.22,55.53。hrms(esi):m/zcalc。forc16h13o3[m+h]+:253.0865,found:253.0865。
实施例五
在15ml单口反应管中,加入23.8mg(0.1mmol)的a5,238mg的sba-sh-15%,于140℃下搅拌。约24h后tlc检测反应完毕,加入5ml氯仿提取,抽滤,取滤液减压旋蒸除去氯仿得到固体,将该固体用甲醇重结晶得产品(b5)19mg,收率82%,白色固体。
b5的谱图数据如下:1hnmr(400mhz,cdcl3)δ8.24(dd,j=7.9,1.3hz,1h),7.82(d,j=8.2hz,2h),7.73–7.66(m,1h),7.57(d,j=8.3hz,1h),7.42(t,j=7.5hz,1h),7.32(d,j=8.1hz,2h),6.80(s,1h),2.44(s,3h)。13cnmr(101mhz,cdcl3)δ178.47,163.57,156.21,142.26,133.67,129.77,128.90,126.21,125.65,125.13,123.97,118.07,106.94,21.56。hrms(esi):m/zcalc。forc16h13o2[m+h]+:237.0916,found:237.0913。
实施例六
在15ml单口反应管中,加入23.0mg(0.1mmol)的a6,230mg的sba-sh-15%,于140℃下搅拌。约24h后tlc检测反应完毕,加入5ml氯仿提取,抽滤,取滤液减压旋蒸除去氯仿得到固体,将该固体用甲醇重结晶得产品(b6)15mg,收率66%,白色固体。
b6的谱图数据如下:1hnmr(400mhz,cdcl3)δ8.22(dd,j=7.9,1.6hz,1h),7.74(dd,j=3.8,1.0hz,1h),7.70(ddd,j=8.6,7.2,1.6hz,1h),7.60(dd,j=5.0,1.0hz,1h),7.54(d,j=8.3hz,1h),7.47–7.39(m,1h),7.20(dd,j=4.9,3.8hz,1h),6.72(s,1h)。13cnmr(101mhz,cdcl3)δ177.91,159.03,155.91,135.15,133.76,130.28,128.51,128.46,125.68,125.28,123.99,117.95,106.20.hrms(esi):m/zcalc。forc13h9o2s[m+h]+:229.0318,found:229.0320。
实施例七
在15ml单口反应管中,加入30.3mg(0.1mmol)的a7,303mgsba-sh-15%介孔材料,于140℃下搅拌。约24h后tlc检测反应完毕,加入5ml氯仿提取,抽滤,取滤液减压旋蒸除去氯仿得到固体,将该固体用甲醇重结晶得产品(b7)26mg,收率87%,白色固体。
b7的谱图数据如下:1hnmr(400mhz,cdcl3)δ8.38(d,j=2.4hz,1h),7.94(dd,j=7.8,1.6hz,2h),7.81(dd,j=8.9,2.4hz,1h),7.63–7.53(m,3h),7.50(d,j=8.9hz,1h),6.86(s,1h).13cnmr(101mhz,cdcl3)δ177.06,163.71,155.02,136.75,131.91,131.40,129.14,128.40,126.35,125.30,120.07,118.70,107.58.hrms(esi):m/zcalc。forc15h10bro2[m+h]+:300.9864,found:300.9867。
实施例八
在15ml单口反应管中,加入28.4mg(0.1mmol)的a8,284mgsba-sh-15%介孔材料,于140℃下搅拌。约24h后tlc检测反应完毕,加入5ml氯仿提取,抽滤,取滤液减压旋蒸除去氯仿得到固体,将该固体用甲醇重结晶得产品(b8)25mg,收率88%,白色固体。
b8的谱图数据如下:1hnmr(400mhz,cdcl3)δ8.14(d,j=8.7hz,1h),7.88(d,j=8.9hz,2h),7.08–6.95(m,4h),6.70(s,1h),3.95(s,3h),3.91(s,3h)。13cnmr(101mhz,cdcl3)δ177.88,164.04,163.05,162.24,157.90,127.85,127.01,124.13,117.79,114.42,114.20,106.11,100.40,55.84,55.51。hrms(esi)m/zcalc。forc17h15o4[m+h]+:283.0970,found:283.0972.
实施例九
在15ml单口反应管中,加入25.4mg(0.1mmol)的a9,230mg的sba-sh-15%,于140℃下搅拌。约24h后tlc检测反应完毕,加入5ml氯仿提取,抽滤,取滤液减压旋蒸除去氯仿得到固体,将该固体用甲醇重结晶得产品(b9)13mg,收率51%,白色固体。
b9的谱图数据如下:1hnmr(400mhz,cdcl3)δ7.94–7.85(m,2h),7.59(t,j=8.4hz,1h),7.55–7.47(m,3h),7.15(d,j=8.4hz,1h),6.84(d,j=8.3hz,1h),6.76(s,1h),4.02(s,3h)。13cnmr(101mhz,cdcl3)δ178.40,161.11,159.78,158.32,133.79,131.46,131.39,128.99,126.08,114.60,110.19,109.11,106.45,56.53。hrms(esi):m/zcalc.forc16h13o3[m+h]+:253.0865,found:253.0865。
实施例十
在15ml单口反应管中,加入30.2mg(0.1mmol)的a10,302mg的sba-sh-15%,于140℃下搅拌。约24h后tlc检测反应完毕,加入5ml氯仿提取,抽滤,取滤液减压旋蒸除去氯仿得到固体,将该固体用甲醇重结晶得产品(b10)12mg,收率41%,白色固体。
b10的谱图数据如下:1hnmr(400mhz,cdcl3)δ8.17(dd,j=6.3,3.4hz,1h),8.04(d,j=8.2hz,1h),7.97(dd,j=6.2,3.3hz,1h),7.83–7.75(m,1h),7.60(tt,j=6.1,5.5hz,4h),7.12(d,j=8.4hz,1h),6.90(d,j=8.3hz,1h),6.63(s,1h),4.06(s,3h)。13cnmr(101mhz,cdcl3)δ178.18,163.15,159.97,158.79,133.89,133.75,131.40,130.42,130.30,129.76,128.72,127.89,127.38,126.53,125.08,124.95,114.57,110.31,106.59,56.59.hrms(esi):m/zcalc。forc20h15o3[m+h]+:303.1016,found:303.1023。
实施例十一
在15ml单口反应管中,加入28.2mg(0.1mmol)的a11,282mg的sba-sh-15%,于140℃下搅拌。约24h后tlc检测反应完毕,加入5ml氯仿提取,抽滤,取滤液减压旋蒸除去氯仿得到固体,将该固体用甲醇重结晶得产品(b11)11mg,收率38%,白色固体。
b11的谱图数据如下:1hnmr(400mhz,cdcl3)δ7.58(t,j=8.4hz,1h),7.45(d,j=7.6hz,1h),7.14(d,j=8.4hz,2h),7.07(d,j=8.4hz,1h),6.86(d,j=8.3hz,1h),6.42(s,1h),4.03(s,3h),2.48(s,3h),2.40(d,j=11.7hz,3h)。13cnmr(101mhz,cdcl3)δ178.32,163.90,159.86,158.58,140.91,136.68,133.69,132.08,129.44,129.15,126.92,114.50,113.21,110.15,106.38,56.53,29.72,20.63.hrms(esi):m/zcalc。forc18h17o3[m+h]+:281.1172,found:281.1186。
实施例十二
在15ml单口反应管中,加入34.4mg(0.1mmol)的a12,344mg的sba-sh-15%,于140℃下搅拌。约24h后tlc检测反应完毕,加入5ml氯仿提取,抽滤,取滤液减压旋蒸除去氯仿得到固体,将该固体用甲醇重结晶得产品(b12)15mg,收率43%,白色固体。
b12的谱图数据如下:1hnmr(400mhz,cdcl3)δ7.59(t,j=8.4hz,1h),7.16(d,j=8.4hz,1h),7.11(s,2h),6.84(d,j=8.3hz,1h),6.70(s,1h),4.01(s,3h),3.96(s,6h),3.93(s,3h)。13cnmr(101mhz,cdcl3)δ178.31,160.90,159.75,158.22,153.51,140.90,133.75,126.65,114.48,110.15,108.85,106.48,103.41,61.03,56.53,56.32.hrms(esi):m/zcalc。forc19h19o6[m+h]+:343.1176,found:343.1185。
实施例十三
在15ml单口反应管中,加入31.4mg(0.1mmol)的a13,314mgsba-sh-15%,于140℃下搅拌。约24h后tlc检测反应完毕,加入5ml氯仿提取,抽滤,取滤液减压旋蒸除去氯仿得到固体,将该固体用甲醇重结晶得产品(b13)17mg,收率55%,白色固体。
b13的谱图数据如下:1hnmr(400mhz,cdcl3)δ7.48(d,j=7.8hz,1h),7.45–7.37(m,2h),7.06(dd,j=8.0,1.5hz,1h),6.69(s,1h),6.59(d,j=2.2hz,1h),6.40(d,j=2.2hz,1h),3.98(s,3h),3.94(s,3h),3.91(s,3h)。13cnmr(101mhz,cdcl3)δ177.66,164.10,160.92,160.47,159.97,159.92,132.90,130.03,118.40,116.91,111.32,109.33,109.30,96.22,92.84,56.46,55.79,55.49.hrms(esi):m/zcalc。forc18h17o5[m+h]+:313.1076,found:313.1077。
实施例十四
在15ml单口反应管中,加入27.3mg(0.1mmol)的a14,273mg的sba-sh-15%,于140℃下搅拌。约24h后tlc检测反应完毕,加入5ml氯仿提取,抽滤,取滤液减压旋蒸除去氯仿得到固体,将该固体用甲醇重结晶得产品(b14)25mg,收率92%,白色固体。
b14的谱图数据如下:1hnmr(400mhz,dmso-d6)δ11.74(s,1h),7.83(dd,j=13.1,8.7hz,3h),7.73(dd,j=9.3,3.0hz,1h),7.59(td,j=8.7,3.1hz,1h),7.15(d,j=8.3hz,2h),6.33(s,1h),3.86(s,3h)。13cnmr(101mhz,dmso)δ176.45,161.63,150.32,137.66,129.35,126.40,121.80,121.08,120.76,114.98,109.30,109.08,106.23,55.93。hrms(esi):m/zcalc.forc16h14fno2[m+h]+:272.1042,found:272.1041。
实施例十五
在15ml单口反应管中,加入28.3mg(0.1mmol)的a15,283mg的sba-sh-15%,于140℃下搅拌。约24h后tlc检测反应完毕,加入5ml氯仿提取,抽滤,取滤液减压旋蒸除去氯仿得到固体,将该固体用甲醇重结晶得产品(b15)24mg,收率87%,白色固体。
b15的谱图数据如下:1hnmr(400mhz,dmso-d6)δ11.73(s,1h),8.11(d,j=7.9hz,1h),7.80(d,j=8.3hz,1h)7.69(t,j=7.76hz,1h),7.49-7.39(m,2h),7.36(t,j=7.5hz,1h),7.16(d,j=8.4hz,1h),6.45(s,1h),3.88(d,j=18.6hz,6h)。13cnmr(400mhz,dmso-d6)δ179.67,151.29,150.68,149.38,140.99,132.24,126.82,125.05,123.79,120.71,119.24,112.30,111.35,106.96,75.05,70.25,56.26.hrms(esi):m/zcalc。forc15h12brno[m+h]+:281.1052,found:282.1085。
- 还没有人留言评论。精彩留言会获得点赞!