一种四嗪类超交联多孔光催化剂的合成方法与流程
本发明涉及有机合成和功能材料技术领域,具体涉及一种四嗪类超交联多孔光催化剂的合成及其在光催化领域中的应用。
背景技术:
面对当前能源和环境危机的挑战,光催化以其温和高效的反应速率以及反应的绿色化而受到越来越多的人关注。上世纪70年代以来,科学工作者已发展了基于钛、镓、镉、锌、钨等元素的各种无机半导体材料(光敏剂或光催化剂)。但是,由于无机物本身结构的限制,这类物质毒性较大、对可见光的吸收范围比较窄、禁带带隙宽,对阳光的利用效率很低,并且在催化有机物的合成时选择性比较差。与此不同的是,卟啉等有机染料种类繁多,具有更加优异的光学活性、适宜的禁带宽度以及易于调节的结构特性。科学工作者根据有机染料在化学键的断裂、重组、编织等过程中发挥的重要作用实现了一系列均相催化剂的简易合成。因此,有机染料的发现和应用大大地推动了光催化的发展。但需要指出的是,这些均相催化剂难以与产物分离、不易回收利用,并且有些价格非常昂贵。因此,人们需要发展出价格便宜,易于制备和调节,可以循环利用,性能优异,能充分利用可见光,禁带带隙适当的多相催化剂,才有可能广泛实现光催化的工业化。
超高交联聚合物(hcps)是一类新兴的由共价键连接而成的有机多孔材料。与传统的无机材料(如分子筛、活性炭)以及其他多孔材料相比,hcps连接方式和功能性单体的种类多,可设计性更强,合成更为简便,比表面积更大,密度小(一般只有c、h、o、n等元素)。hcps具有刚性的结构以及大π共轭体系,这为孔的形成、催化位点的暴露、光的吸收以及电子的转移提供了结构基础。另外,由于是通过强共价键连接形成的,hcps也比一些有机-无机杂化材料(如有序介孔有机硅和金属有机框架)具有更好的稳定性。此外,其紫外-可见光吸收范围一般较无机半导体宽,禁带带隙较窄,可见光利用率较高。因此,结合其光电性质以及其多相催化的性能,hcps作为光催化剂具独特的优势。
1,2,4,5-四嗪单元可以看作是苯环上的4个次甲基(-ch=)被4个叔氨基(-n=)取代之后杂环化合物,其分子式为c2h2n4,氮元素所占质量分数达68.3%,碳氢元素质量分数分别是29.3%和2.5%,是一种典型的高氮杂环化合物。氮的电负性较大,环上电子云偏向氮,在诱导效应的作用下,环上缺电子程度较大,碳原子带有较多的正电荷,有利于发生亲核取代反应。同时,该四嗪单元具有良好的刚性以及大π共轭体系,有利于构建有机多孔材料。综合以上特点,将四嗪单元引入hcps中,合成以四嗪单元为前体的hcps材料,在功能化hcps材料方面具有重要意义。
综上,本发明提供了一种四嗪类超交联多孔光催化剂的合成方法,实现了将四嗪单元引入到hcps材料中,并运用所得到的材料合成了苯并咪唑类化合物。
技术实现要素:
为了克服hcps在功能化应用方面的难题,本发明的一个目的是提供了一种四嗪类超交联多孔光催化剂的合成方法。所采用的具体技术方案如下:
一种四嗪类超交联多孔光催化剂的合成方法,包括以下步骤:将前体甲氧基取代的双吡啶四嗪单体、芳香砌块,以及催化剂三氯化铁置于干燥的两口烧瓶内,氮气保护下加入溶剂1,2-二氯乙烷和交联剂二甲氧基甲烷,45℃低温搅拌5~24h后,再升温至90℃继续反应19~48h,反应结束后经过甲醇和浓盐酸处理、真空抽滤、索氏提取、干燥即可制得四嗪类超交联多孔光催化剂tz-hcps。
进一步地,甲氧基取代的双吡啶四嗪单体和芳香砌块的摩尔比为1:0-50,优选甲氧基取代的双吡啶四嗪单体和芳香砌块的摩尔比为1:20。
进一步地,所述前体浓度:甲氧基取代的双吡啶四嗪单体的浓度为0.02~0.40mmol/ml,芳香砌块的浓度为0.0~1.00mmol/ml。
进一步地,所述芳香砌块包括但不限于苯、芴。
进一步地,所述二甲氧基甲烷和1,2-二氯乙烷的体积比为1:0-20,优选二甲氧基甲烷和1,2-二氯乙烷体积比为1:6。
进一步地,所述反应温度为30~150℃,优选为45~90℃;反应时间为1~5d,优选为1~3d。
优选地,加热操作具体为45℃低温搅拌5h后,再升温至90℃继续反应19h。
基于甲氧基取代的双吡啶四嗪单体合成的tz-hcp类催化剂,其纽节点单元包含但不仅限于苯、芴等芳香烃。
本发明的另一个目的是提供了采用上述合成方法制备得到的四嗪类超交联多孔光催化剂。
本发明还提供了所述四嗪类超交联多孔光催化剂在催化光反应方面的应用。
进一步地,本发明提供了四嗪类超交联多孔光催化剂在催化邻苯二胺与醛类亲电试剂间的偶联反应方面的应用。
通过本发明中所述的合成方法得到的四嗪类超交联多孔光催化剂可用于光催化邻苯二胺与对氯苯甲醛等亲电试剂(如苯甲醛、丙醛、1-芘甲醛等)间的偶联反应,实现了苯并咪唑类化合物的合成。
本发明提供的四嗪类超交联多孔光催化剂的合成方法,与现有技术相比具有以下有益效果:
1.本发明提供的合成方法得到的tz-hcps催化剂密度小(仅含碳、氢、氧、氮元素),并且具有较大的比表面积以及宽的可见光吸收范围;
2.本发明制得的tz-hcps催化剂在室温、空气氛围以及可见光光照下可以高效地催化邻苯二胺与对氯苯甲醛等亲电试剂(如苯甲醛、丙醛、1-芘甲醛等)间的偶联反应,实现了苯并咪唑类化合物的合成,而且具有良好的重复使用性,至少可以催化使用20次。
附图说明
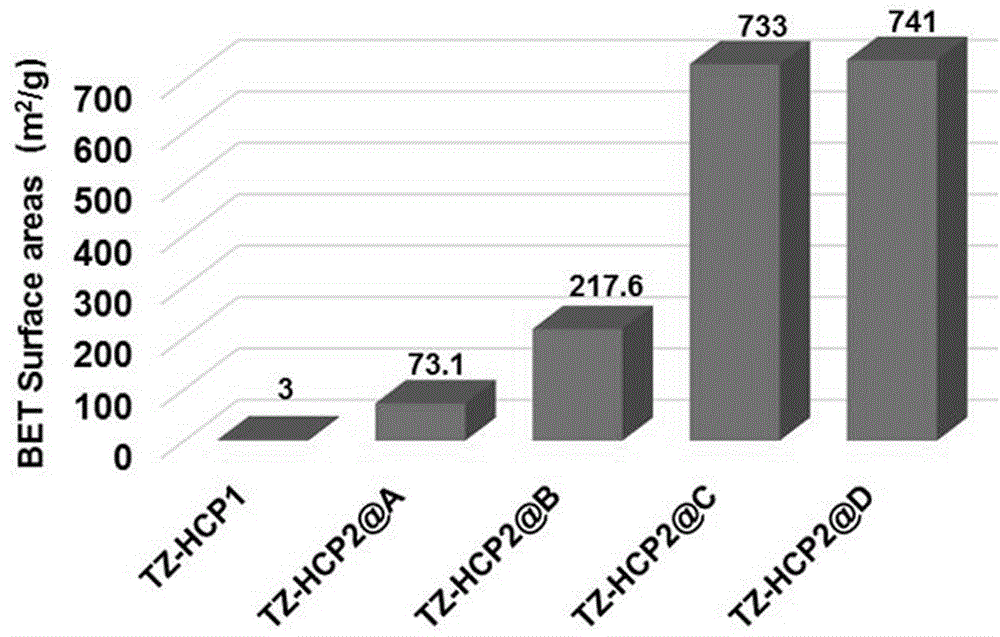
图1是本发明所合成的tz-hcp1、tz-hcp2@a、tz-hcp2@b、tz-hcp2@c及tz-hcp2@d的bet比表面积测试结果。
图2是本发明所合成的tz-hcp2@b、tz-hcp2@c、tz-hcp2@d及tz-hcp3的氮气吸脱附曲线。
图3是本发明所合成的tz-hcp2@b、tz-hcp2@c、tz-hcp2@d及tz-hcp3的孔径分布曲线。
图4是本发明所合成的tz-hcp2@d及tz-hcp3固体核磁谱图。
图5是本发明所合成的tz-hcp2@c、tz-hcp2@d、tz-hcp3以及原料(iii)的傅里叶红外图谱,其中(iii)为甲氧基取代的双吡啶四嗪。
图6是本发明所合成的tz-hcp2@b、tz-hcp2@c、tz-hcp2@d及tz-hcp3的热分析曲线。
图7是本发明所合成tz-hcp2@d的催化测试。
图8是本发明所合成tz-hcp2@d的循环测试。
具体实施方式
为更进一步阐述本发明所采取的技术手段及其效果,以下结合附图及本发明的优选实施例进行详细描述。
(1)材料前驱体的合成
合成路线如式(ⅰ)所示:
本发明所用的底物苯或芴和二甲氧基甲烷均为市场可购产品。甲氧基取代的双吡啶四嗪单体可由5-(4-甲氧基苯)-2-氰基吡啶制备,5-(4-甲氧基苯)-2-氰基吡啶(acsomega2017,2,5666-5683.)采用文献报道的方法通过简单转化,可比较简易地得到。合成方法具体如下:
第一步:将对甲氧基苯硼酸(996mg,6.55mmol),5-溴-2-氰基吡啶(1g,5.46mmol),碳酸钾(2.26g,16.38mmol)以及四(三苯基磷)钯(500mg,0.43mmol)置于干燥的烧瓶中,然后往烧瓶中加入40ml四氢呋喃和20ml水溶液,氮气保护下80℃搅拌5-24h,之后真空旋除四氢呋喃,经过二氯甲烷(100ml)萃取,柱色谱纯化得到淡黄色固体(i),产率98%。
第二步:将5-(4-甲氧基苯)-2-氰基吡啶(371.4mg,1.76mmol)和硫粉(28mg,0.88mmol)置于干燥的烧瓶中并加入2ml乙醇,90℃搅拌加热,回流状态下加入1ml水合肼,然后继续回流加热5-24h。最后,将反应体系冷却至0℃并抽滤,同时用-30℃的冰乙醇(20ml)洗涤,可得到棕黄色固体(ii)。
第三步:将固体(ii)分散至4ml的冰乙酸中,室温搅拌下分批加入亚硝酸钠(0.40g,5.79mmol),亚硝酸钠加入过程中有棕红色气体生成,加毕,室温下继续搅拌5-24h。将反应体系倒入到饱和碳酸钾溶液(100ml)中,静置、抽滤、大量水洗,烘箱内50℃干燥得到紫红色固体(iii),产率34%。
(2)四嗪类超交联多孔光催化剂(tz-hcp)的合成
反应式如下式(ⅱ)所示:
实施例1
将甲氧基取代的双吡啶四嗪单体(165mg,0.37mmol)和三氯化铁(2.4g,14mmol)置于干燥的两口烧瓶中,氮气保护下抽真空3-5次,然后通过针管向烧瓶中加入7.4ml1,2-二氯乙烷,室温搅拌0.5h,再向烧瓶中加入1.33ml二甲氧基甲烷,45℃低温搅拌5h后,升温至90℃继续反应19h。然后向烧瓶中加入20ml甲醇,90℃搅拌0.5h,真空抽滤,经n,n-二甲基甲酰胺、甲醇洗涤后,将固体转移至烧瓶中,加入20ml浓盐酸,室温搅拌2h,真空抽滤,经n,n-二甲基甲酰胺、甲醇、二氯甲烷依次洗涤后,对残渣进行48h索氏提取(甲醇:二氯甲烷=1:1),烘箱干燥即可制得四嗪类超交联多孔光催化剂tz-hcp1(棕黑色固体粉末)568mg。
实施例2
将甲氧基取代的双吡啶四嗪单体(179mg,0.4mmol)和三氯化铁(520mg,3.2mmol)置于干燥的两口烧瓶中,氮气保护下抽真空3-5次,然后向烧瓶中加入1.6ml1,2-二氯乙烷和0.11ml苯,室温搅拌0.5h,再向烧瓶中加入0.29ml二甲氧基甲烷,45℃低温搅拌5h后,升温至90℃继续反应19h。将反应体系真空抽滤,经石油醚、丙酮依次洗涤后,将固体转移至干燥的烧瓶中,加入20ml盐酸(6m),室温搅拌2h。真空抽滤,经n,n-二甲基甲酰胺、甲醇、二氯甲烷依次洗涤后,以丙酮为溶剂对粗产品进行索氏提取48h,烘箱中80℃干燥即可制得四嗪类超交联多孔光催化剂tz-hcp2@a(黑色固体粉末)241mg。
实施例3
将甲氧基取代的双吡啶四嗪单体(103mg,0.23mmol)和三氯化铁(464mg,2.86mmol)置于干燥的两口烧瓶中,氮气保护下抽真空3-5次,然后向烧瓶中加入1.43ml1,2-二氯乙烷和0.11ml苯,室温搅拌0.5h,再向烧瓶中加入0.26ml二甲氧基甲烷,45℃低温搅拌5h后,升温至90℃继续反应19h。将反应体系真空抽滤,经石油醚、丙酮依次洗涤后,将固体转移至干燥的烧瓶中,加入20ml盐酸(6m),室温搅拌2h。真空抽滤,经n,n-二甲基甲酰胺、甲醇、二氯甲烷依次洗涤后,以丙酮为溶剂对粗产品进行索氏提取48h,烘箱中80℃干燥即可制得四嗪类超交联多孔光催化剂tz-hcp2@b(棕色固体粉末)214mg。
实施例4
将甲氧基取代的双吡啶四嗪单体(165mg,0.37mmol)和三氯化铁(2.40g,14mmol)置于干燥的两口烧瓶中,氮气保护下抽真空3-5次,然后向烧瓶中加入7.4ml1,2-二氯乙烷和0.66ml苯,室温搅拌0.5h,再向烧瓶中加入1.33ml二甲氧基甲烷,45℃低温搅拌24h后,补加7.4ml1,2-二氯乙烷。升温至90℃继续反应48h。将反应体系真空抽滤,经石油醚、丙酮依次洗涤后,将固体转移至干燥的烧瓶中,加入20ml盐酸(6m),室温搅拌2h。真空抽滤,经n,n-二甲基甲酰胺、甲醇、二氯甲烷依次洗涤后,以丙酮为溶剂对粗产品进行索氏提取48h,烘箱中80℃干燥即可制得四嗪类超交联多孔光催化剂tz-hcp2@c(红棕色固体粉末)977mg。
实施例5
将甲氧基取代的双吡啶四嗪单体(165mg,0.37mmol)和三氯化铁(2.4g,14mmol)置于干燥的两口烧瓶中,氮气保护下抽真空3-5次,然后通过针管向烧瓶中加入7.4ml1,2-二氯乙烷和0.66ml苯,室温搅拌0.5h,再向烧瓶中加入1.33ml二甲氧基甲烷,45℃低温搅拌5h后,升温至90℃继续反应19h。然后向烧瓶中加入20ml甲醇,90℃搅拌0.5h,真空抽滤,经n,n-二甲基甲酰胺、甲醇洗涤后,将固体转移至烧瓶中,加入20ml浓盐酸,室温搅拌2h,真空抽滤,经n,n-二甲基甲酰胺、甲醇、二氯甲烷依次洗涤后,对残渣进行48h索氏提取(甲醇:二氯甲烷=1:1),烘箱干燥即可制得四嗪类超交联多孔光催化剂tz-hcp2@d(红棕色固体粉末)880mg。
实施例6
将甲氧基取代的双吡啶四嗪单体(82.50mg,0.18mmol),芴(612mg,3.68mmol)和三氯化铁(1.20g,7.38mmol)置于干燥的两口烧瓶中,氮气保护下抽真空3-5次,然后通过针管向烧瓶中加入3.7ml1,2-二氯乙烷,室温搅拌0.5h,再向烧瓶中加入0.67ml二甲氧基甲烷,45℃低温搅拌5h后,升温至90℃继续反应19h。然后向烧瓶中加入20ml甲醇,90℃搅拌0.5h,真空抽滤,经水、饱和碳酸氢钠溶液、n,n-二甲基甲酰胺、甲醇依次洗涤后,将固体转移至烧瓶中。加入20ml浓盐酸,室温搅拌2h,真空抽滤,经n,n-二甲基甲酰胺、甲醇、二氯甲烷依次洗涤后,对残渣进行24h索氏提取(甲醇:二氯甲烷=1:1),烘箱干燥即可制得四嗪类超交联多孔光催化剂tz-hcp3(土黄色固体粉末)798mg。
如图1所示,不同的实施方法所获得的tz-hcp1和tz-hcp2材料的bet比表面积不同。tz-hcp1、tz-hcp2@a、tz-hcp2@b、tz-hcp2@c以及tz-hcp2@d的比表面积分别为3.0m2/g、73.1m2/g、217.6m2/g、733.0m2/g、741.0m2/g,说明单体(iii)的含量越多,bet比表面积越小。其中,tz-hcp2@d比表面积最大,合成所耗时间最短。
如图2,通过氮气吸脱附曲线可知tz-hcp2@b、tz-hcp2@c、tz-hcp2@d以及tz-hcp3对氮气的吸附量不同。tz-hcp2@c、tz-hcp2@d以及tz-hcp3表现出较好的吸附效果。
如图3,通过nldft方法计算得到tz-hcp2@b、tz-hcp2@c、tz-hcp2@d以及tz-hcp3的孔径分布,其中tz-hcp2@b孔径主要分布在4.1nm左右,tz-hcp2@c孔径主要分布在4.2nm左右,tz-hcp2@d孔径主要分布在4.2nm左右,tz-hcp3的孔径主要分布在4.1nm左右。
如图4,通过固体核磁碳谱可以看到55ppm、158ppm、36(37)ppm等信号,它们可分别被归属为材料中的甲氧基、亚胺键以及交联后形成的亚甲基。这说明通过傅克烷基化反应以及亚甲基连接,双吡啶四嗪单体被成功地嵌入到了有机多孔材料中。
如图5,通过对比tz-hcp和原料的傅里叶红外谱图,可以发现tz-hcp的红外谱图与原料(iii)有相似之处,都有明显的c=n键振动峰,说明四嗪单元被成功地引入到了hcp材料中。
如图6,通过热分析可以发现,tz-hcp2@b在氮气气氛下可以稳定到295℃而不发生明显的分解,tz-hcp2@c在氮气气氛下可以稳定到318℃而不发生明显的分解(低于100℃的分解来自于材料中的空气、水蒸气等易挥发物),tz-hcp2@d在氮气气氛下可以稳定到319℃而不发生明显的分解,tz-hcp3在氮气气氛下可以稳定到335℃而不发生明显的分解。
(3)tz-hcp2@d的催化测试
实施例7
反应式如下式(ⅲ)所示:
将对氯苯甲醛(0.20mmol)等亲电试剂(如苯甲醛、丙醛、1-芘甲醛等),邻苯二胺(21.6mg,0.20mmol)和tz-hcp2@d(10mg)加入到10ml反应管中,加入2ml乙醇,然后于6wled白灯光照条件下室温搅拌0.5-4h。将反应体系用丙酮离心洗涤(5×5ml),合并有机相,真空旋除溶剂,残余物通过柱色谱分离即可得到产物。
如图7,tz-hcp2@d催化邻苯二胺和对氯苯甲醛等亲电试剂(如苯甲醛、丙醛、1-芘甲醛等)间的偶联反应产率大部分在90%以上,可以看出,tz-hcp2@d具有良好的光催化活性。
(4)tz-hcp2@d的循环测试:
实施例8
反应式如下式(ⅳ)所示:
将邻苯二胺(21.6mg,0.20mmol)和tz-hcp2@d(10mg)加入10ml反应管中,加入2ml乙醇,加入20.4μl苯甲醛(0.20mmol),然后于6wled白灯光照条件下室温搅拌0.5-2h。将反应体系用丙酮离心洗涤(5×5ml),合并有机相,真空旋除溶剂,残余物通过柱色谱分离即可得到产物。
如图8,tz-hcp2@d催化邻苯二胺和苯甲醛的偶联反应重复20次,每次反应时间1.0-2.5h,偶联反应产物的产率均能保持在95-99%。由此可以看出,重复使用对tz-hcp2@d的催化效果影响不大,说明tz-hcp2@d具有良好的催化稳定性。
以上所述,仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,本领域普通技术人员对本发明的技术方案所做的其他修改或者等同替换,只要不脱离本发明技术方案的精神和范围,均应涵盖在本发明的权利要求范围当中。
- 还没有人留言评论。精彩留言会获得点赞!