一种改性氮化碳/Fe基MOF复合材料及其制备方法和应用与流程
本发明涉及可见光催化技术领域,具体涉及一种改性氮化碳/fe基mof复合材料及其制备方法和应用。
背景技术:
以重金属铬为例,制革、电镀等行业每年产生大量的含铬cr(vi)有机废水,形成的复合污染难治理,毒性增强。所以如何有效去除水体中的cr(vi)是一个亟待解决的问题。
传统的化学还原法需要分步处理,且产生大量含铬污泥。而光催化作为一种清洁的环境友好型技术,由于能在光照下产生电子空穴对,兼具氧化还原性,可以一步处理含cr(vi)复合物。但是目前研究较多的tio2等半导体催化剂仍然存在一些不足之处,比如比表面积较小、带隙较高、光响应能力较差等问题,亟需开发新型、高效可见光催化剂。
金属有机框架材料(metal-organicframeworks,mof)是由无机金属中心(金属离子或金属簇)与桥连的有机配体通过自组装相互连接,形成的一类具有周期性网络结构的晶态多孔材料。mofs材料的优势在于其具有较大的比表面积和极高的孔隙率,在传感、吸附、药物等领域有着巨大的潜力而受到人们的广泛关注,同时mofs也是一种具有催化性质的材料。从1999年mof-5作为的第一种在实验室被合成的mofs材料后发展十分迅速,已经有了成千上万种mofs材料被报道出来。
近年来,mofs在光催化领域展现较好的应用前景,尤其以铁离子为金属中心的fe-mofs,如mil-53(fe)、mil-101(fe)、mil-88(fe)等,这些mofs催化剂的共同点是在无机金属中心中引入了有机羧酸基团,形成了性质稳定、比表面积大、多空的三维空间结构mofs材料。由于mofs材料在结构上和组成上具有多样性,同时可调性较强,因此可根据实际的需求来设计mofs的结构并对其进行改性。
技术实现要素:
针对本领域存在的不足之处,本发明提供了一种改性氮化碳/fe基mof复合材料的制备方法,采用“前功能化”的方法,以改性氮化碳(记为g-c3n4-m)作为改性物质,制备了新型改性氮化碳/fe基mof复合材料(记为g-c3n4-m/nh2-mil-53(fe)-x%),可作为光催化剂,可用于cr(vi)的可见光催化还原。性能测试表明,添加了极少量改性氮化碳g-c3n4-m的nh2-mil-53(fe)-x%在可见光下对于cr(vi)的还原效率明显优于传统g-c3n4构成的复合材料g-c3n4/nh2-mil-53(fe)-x%以及单一nh2-mil-53(fe)-x%和单一g-c3n4-m,说明g-c3n4-m和nh2-mil-53(fe)-x%之间存在强烈的协同作用。
一种改性氮化碳/fe基mof复合材料的制备方法,包括步骤:
(1)将三聚氰胺和三氨基嘧啶按摩尔比1:2~4混合均匀后,在惰性气氛下450~550℃焙烧,将所得产物研磨后加入二甲基亚砜(dmso)中,超声剥离1~3h后,用蒸馏水离心、洗涤、干燥得到改性氮化碳粉末;
(2)将步骤(1)得到的改性氮化碳粉末分散于n,n-二甲基甲酰胺(dmf)中形成分散液,将所述分散液加入到含有氯化铁、对苯二甲酸和氨基对苯二甲酸的n,n-二甲基甲酰胺溶液中,混匀后于140~160℃溶剂热反应14~16h,得到改性氮化碳/fe基mof复合材料。
本发明制备过程工艺简单且反应条件较为温和,所得复合材料为g-c3n4-m/nh2-mil-53(fe)-x%,可作为光催化剂。本发明首先以氨基对苯二甲酸、对苯二甲酸混合配体制备异质结型nh2-mil-53/mil-53(即nh2-mil-53(fe)-x%,x%代表氨基化比例),发现mil-53的氨基化显著影响材料的循环稳定性,且氨基化的比例同样影响光催化活性的变化。然后在此优化基础上,本发明制备p型半导体改性氮化碳g-c3n4-m(普通g-c3n4为n型半导体),进一步研究发现g-c3n4-m导带和价带与氨基部分功能化后的fe基mofnh2-mil-53(fe)-x%的导带和价带相匹配,可形成p-n型异质结,从而可以进一步发挥两者在光催化的协同作用,有效提高光催化还原cr(vi)的效率。本发明制备的复合材料具有高光催化活性、合成方便、成本低廉、易回收等优点。
所述惰性气氛为n2、稀有气体等。
作为优选,步骤(1)中,所述焙烧时间为1~3h。
作为优选,步骤(2)中,所述氯化铁、对苯二甲酸和氨基对苯二甲酸的摩尔比为2:0.5~1.5:0.5~1.5,且所述氯化铁摩尔量与对苯二甲酸、氨基对苯二甲酸摩尔量之和的比例为1:1。
作为优选,步骤(2)中,所述分散液中的改性氮化碳粉末与所述氯化铁的比例为0.5~2mg:2mmol。nh2-mil-53(fe)-x%与极少量的改性氮化碳复合后,光催化性能便可以得到显著提升。
本发明还提供了所述的制备方法制备得到的改性氮化碳/fe基mof复合材料,可作为光催化剂。
本发明还提供了所述的改性氮化碳/fe基mof复合材料在光催化领域中的应用。例如所述的改性氮化碳/fe基mof复合材料可作为光催化剂,或者用于制备光催化剂。
本发明还提供了一种含cr(vi)废水的处理方法,包括步骤:将所述的改性氮化碳/fe基mof复合材料加入所述含cr(vi)废水中,暗反应吸附平衡后,进行可见光照射,进行光催化降解。
作为优选,暗反应吸附前,向所述含cr(vi)废水中还加入草酸铵,加入量为20~40mg/l。
本发明与现有技术相比,主要优点包括:本发明提供的合成方法工艺简单易行,条件温和,比较适合大规模生产。所得复合材料g-c3n4-m/nh2-mil-53(fe)可作为光催化剂。不同于普通g-c3n4,本发明制备的g-c3n4-m为p型半导体,由于其导带和价带与氨基部分功能化后的fe基mofnh2-mil-53(fe)-x%的导带和价带匹配,可形成p-n型异质结,充分发挥两者在光催化的协同作用,从而可以有效提高光催化还原cr(vi)的效率。本发明制备的复合材料具有高光催化活性、合成方便、成本低廉、易回收等优点。
附图说明
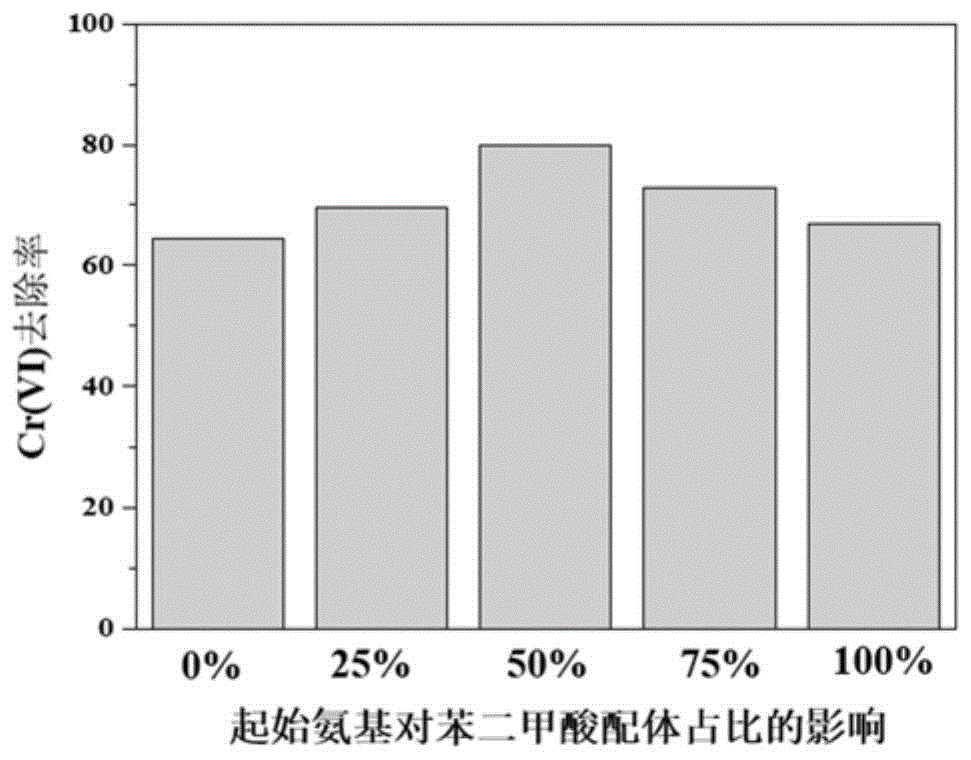
图1为实施例1中以起始氨基对苯二甲酸、对苯二甲酸不同比例制备的nh2-mil-53(fe)-x%的光催化还原cr(vi)性能对比图;
图2为实施例3中起始g-c3n4-m剥离液添加体积对g-c3n4-m/nh2-mil-53(fe)-50%的光催化性能影响图;
图3为实施例4中不同种类催化剂光催化还原cr(vi)的性能对比,其中(1)g-c3n4,(2)g-c3n4-m,(3)mil-53(fe),(4)nh2-mil-53(fe),(5)nh2-mil-53(fe)-50%,(6)g-c3n4/nh2-mil-53(fe)-50%-1.5ml,(7)g-c3n4-m/nh2-mil-53(fe)-50%-1.5ml;
图4为实施例5优化制备的g-c3n4-m/nh2-mil-53(fe)-50%-1.5ml处理含铬废水的循环稳定性图。
具体实施方式
下面结合附图及具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的操作方法,通常按照常规条件,或按照制造厂商所建议的条件。
实施例1
含不同比例氨基对苯二甲酸配体的nh2-mil-53(fe)-x%制备
1)nh2-mil-53(fe)-100%(即nh2-mil-53(fe))的制备
首先,将2mmolfecl3·6h2o和2mmol氨基对苯二甲酸溶解于40ml的n,n-二甲基甲酰胺中,磁力搅拌60min,然后转移到水热釜,在150℃下加热15h,自然冷至室温,以8000rpm速率,离心5min,然后再分别用dmf和甲醇各洗两遍,最后在100℃下真空干燥12h,得所述的nh2-mil-53(fe)-100%。
2)nh2-mil-53(fe)-75%的制备
首先,将2mmolfecl3·6h2o和1.5mmol氨基对苯二甲酸、0.5mmol对苯二甲酸溶解于40ml的n,n-二甲基甲酰胺中,磁力搅拌60min,然后转移到水热釜,在150℃下加热15h,自然冷至室温,以8000rpm速率,离心5min,然后再分别用dmf和甲醇各洗两遍,最后在100℃下真空干燥12h,得所述的nh2-mil-53(fe)-75%。
3)nh2-mil-53(fe)-50%的制备
首先,将2mmolfecl3·6h2o和1mmol氨基对苯二甲酸、1mmol对苯二甲酸溶解于40ml的n,n-二甲基甲酰胺中,磁力搅拌60min,然后转移到水热釜,在150℃下加热15h,自然冷至室温,以8000rpm速率,离心5min,然后再分别用dmf和甲醇各洗两遍,最后在100℃下真空干燥12h,得所述的nh2-mil-53(fe)-50%。
4)nh2-mil-53(fe)-25%的制备
首先,将2mmolfecl3·6h2o和0.5mmol氨基对苯二甲酸、1.5mmol对苯二甲酸溶解于40ml的n,n-二甲基甲酰胺中,磁力搅拌60min,然后转移到水热釜,在150℃下加热15h,自然冷至室温,以8000rpm速率,离心5min,然后再分别用dmf和甲醇各洗两遍,最后在100℃下真空干燥12h,得所述的nh2-mil-53(fe)-25%。
5)nh2-mil-53(fe)-0%(即mil-53(fe))的制备
首先,将2mmolfecl3·6h2o和2mmol对苯二甲酸溶解于40ml的n,n-二甲基甲酰胺中,磁力搅拌60min,然后转移到水热釜,在150℃下加热15h,自然冷至室温,以8000rpm速率,离心5min,然后再分别用dmf和甲醇各洗两遍,最后在100℃下真空干燥12h,得所述的nh2-mil-53(fe)-0%
6)光催化还原cr(vi)性能测试
取10mg上述含不同比例氨基对苯二甲酸配体的nh2-mil-53(fe)-x%,加入到cr(vi)浓度为80μmol/l的重铬酸钾溶液(100ml)中,添加3mg草酸铵作为共存有机物兼空穴捕获剂,调节ph为4.7,在磁力搅拌条件下,暗反应30分钟后,打开氙灯光源,在可见光作用下进行光催化反应。可见光照射20分钟后,取样,离心分离,取上清液用显色法测得其在540nm处的吸光度,通过吸光度在反应前后的对比,可以计算出cr(vi)的还原率,结果如图1所示。
由图1可知,比较不同氨基化比例的nh2-mil-53(fe)-x%对于光催化还原cr(vi)的性能,起始氨基对苯二甲酸和对苯二甲酸的摩尔比例为1:1制备得到的nh2-mil-53(fe)-50%呈现最佳的光催化性能。mil-53(fe)全部氨基化后的光催化性能不如部分氨基化,可见本发明方法原位制备的nh2-mil-53(fe)和mil-53(fe)之间存在协同作用,促进光催化性能提升。
此外,实验过程中发现,在加入草酸铵的情况下,不含氨基的mil-53(fe)稳定性差,易溶解,无法有效分离回收,而含氨基的nh2-mil-53(fe)及其复合物稳定性良好,可循环稳定使用。
实施例2
g-c3n4-m分散液(剥离液)的制备
1)g-c3n4-m的制备
称取三聚氰胺和三氨基嘧啶以摩尔比1:3的量混合均匀后,平铺在石英舟上,在氮气的氛围下在管式炉中加热,以5℃/min的升温速率,升温至500℃后维持120min,最后在氮气吹扫下冷却至室温,得到粉状的g-c3n4-m,研磨成粉末后,在纯dmso中超声处理120min,用来剥离g-c3n4-m颗粒,在离心机内用蒸馏水以8000rpm离心五次后在真空干燥箱内60℃干燥12小时后收集。
2)g-c3n4-m分散液制备
称取步骤1)制备的好的g-c3n4-m颗粒50mg,将其加入到dmf中超声半小时后得到浓度为1mg/ml的g-c3n4-m分散液。
实施例3
多元复合催化剂的制备
1)g-c3n4-m/nh2-mil-53(fe)-50%的制备
首先,将2mmolfecl3·6h2o、1mmol氨基对苯二甲酸、1mmol对苯二甲酸溶解于40ml的n-n-二甲基甲酰胺中,加入不同体积(0.5ml、1.0ml、1.5ml、2ml)实施例2制备的g-c3n4-m分散液,磁力搅拌60min,然后转移到水热釜,在150℃下加热15h,自然冷至室温,以8000rpm速率,离心5min,然后再分别用dmf和甲醇各洗两遍,最后在100℃下真空干燥12h,得g-c3n4-m不同负载比例的g-c3n4-m/nh2-mil-53(fe)-50%。
2)光催化还原cr(vi)性能测试
取10mgg-c3n4-m不同负载比例的g-c3n4-m/nh2-mil-53(fe)-50%,加入到cr(vi)浓度为80μmol/l的重铬酸钾溶液(100ml)中,添加3mg草酸铵作为共存有机物兼空穴捕获剂,调节ph为4.7,在磁力搅拌条件下,暗反应30分钟后,打开氙灯光源,在可见光作用下进行光催化反应。可见光照射20分钟后,取样,离心分离,取上清液用显色法测得其在540nm处的吸光度,通过吸光度在反应前后的对比,可以计算出cr(vi)的还原率,结果如图2所示。
由图2可知,比较g-c3n4-m分散液添加量对于光催化还原cr(vi)的性能,起始添加1.5mlg-c3n4-m剥离液制备得到的g-c3n4-m/nh2-mil-53(fe)-50%-1.5ml呈现最佳的光催化性能。
实施例4
其他对照组催化剂的制备
1)g-c3n4/nh2-mil-53(fe)-50%-1.5ml的制备
首先,将2mmolfecl3·6h2o、1mmol氨基对苯二甲酸、1mmol对苯二甲酸溶解于40ml的n-n-二甲基甲酰胺中,加入1.5mlg-c3n4剥离液(除不添加三氨基嘧啶外,其余制备流程同实施例2的g-c3n4-m剥离液),磁力搅拌60min,然后转移到水热釜,在150℃下加热15h,自然冷至室温,以8000rpm速率,离心5min,然后再分别用dmf和甲醇各洗两遍,最后在100℃下真空干燥12h,得g-c3n4/nh2-mil-53(fe)-50%。
2)g-c3n4的制备
称取三聚氰胺平铺在石英舟上,在氮气的氛围下在管式炉中加热,以5℃·min-1的升温速率,升温至500℃后维持120min,最后在氮气吹扫下冷却至室温,得到粉状的g-c3n4,研磨成粉末后,在纯dmso中超声波浴中处理120min,用来剥离g-c3n4颗粒,在离心机内用蒸馏水以8000rpm离心五次后在真空干燥箱内60℃干燥12小时后收集。
3)各种类催化剂光催化还原cr(vi)性能测试
取10mg不同催化剂,加入到cr(vi)浓度为80μmol/l的重铬酸钾溶液(100ml)中,添加3mg草酸铵作为共存有机物兼空穴捕获剂,调节ph为4.7,在磁力搅拌条件下,暗反应30分钟后,打开氙灯光源,在可见光作用下进行光催化反应。可见光照射20分钟后,取样,离心分离,取上清液用显色法测得其在540nm处的吸光度,通过吸光度在反应前后的对比,可以计算出cr(vi)的还原率,结果如图3所示。
由图3可知,对于光催化还原cr(vi),当以nh2-mil-53(fe)-50%为基底(起始氨基对苯二甲酸与对苯二甲酸摩尔比1:1)、以g-c3n4-m剥离液为添加剂(添加量1.5ml),制备的多元复合催化剂g-c3n4-m/nh2-mil-53(fe)-50%呈现最佳的性能。
本发明g-c3n4-m光催化活性低于普通g-c3n4,且二者均几乎没有光催化活性,但当将极少量g-c3n4-m与部分氨基化的mil-53(fe)复合时,所得复合材料的光催化活性可以得到显著提升,且明显高于普通g-c3n4和部分氨基化的mil-53(fe)复合所得材料,说明g-c3n4-m与部分氨基化的mil-53(fe)形成了多元异质结,存在强烈的协同作用。
实施例5
循环使用稳定性测试
以优化制备的g-c3n4-m/nh2-mil-53(fe)-50%为光催化剂,应用于光催化还原cr(vi),进行5次循环,每次循环前将光催化剂离心、水洗、烘干后重新放入新的80μmol/l的cr(vi)(100ml)溶液中,添加3mg草酸铵作为共存有机物兼空穴捕获剂,调节ph为4.7,在磁力搅拌条件下,暗反应30分钟后,打开氙灯光源,在可见光作用下进行光催化反应。可见光照射20分钟后,取样,离心分离,取上清液用显色法测得其在540nm处的吸光度,通过吸光度在反应前后的对比,可以计算出cr(vi)的还原率,结果如图4所示,g-c3n4-m/nh2-mil-53(fe)-50%具有较好的循环使用稳定性,循环使用5次后其活性并未发生明显变化,且稳定在90%以上。
此外应理解,在阅读了本发明的上述描述内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
- 还没有人留言评论。精彩留言会获得点赞!