一种共轭微孔聚合物/钯镍双金属催化剂的制备方法与流程
本发明属于共轭微孔聚合物制备技术领域,具体涉及一种共轭微孔聚合物/钯镍双金属催化剂的制备方法。
背景技术:
手性共轭微孔聚合物(ccmps)具有简单高效的合成方式、多策略的构筑手段、易与功能化及广泛的应用性等优点,是进一步发展为生产光学纯有机分子的实用催化剂极好的载体。
目前,金属纳米粒子的制备方法主要有浸渍法、溶胶凝胶法、化学还原法等,以浸渍法研究居多。浸渍法虽操作简单,但存在金属纳米粒子稳定性差、颗粒尺寸较大、易发生团聚及能耗大等问题。伴随着负载型手性催化剂研究及应用领域的逐渐推广,出现了一些新问题,即在负载型手性催化剂的使用过程中,成本较高、催化效率较低、催化剂回收困难等问题。
综上可知,探索一种成本低、稳定性好、催化活性高、可回收且制备简便的负载型手性共轭微孔聚合物(ccmps)/钯镍双金属纳米粒子催化剂具有重要的研究意义和科学价值。
技术实现要素:
鉴于此,本发明的目的在于提供一种共轭微孔聚合物/钯镍双金属催化剂的制备方法,以有效解决上述背景技术中提出的问题。
为实现上述目的,本发明提供如下技术方案:
一种共轭微孔聚合物/钯镍双金属催化剂的制备方法,以粉末状含有酰胺键的手性共轭微孔聚合物和乙二醇为原料,超声分散,制得溶液a;以pd(oac)2、ni(oac)2·4h2o、氯仿和去离子水为原料,搅拌混合,制得溶液b;将溶液a与溶液b混合,并通过硼氢化钠还原及室温水浴反应制得共轭微孔聚合物/钯镍双金属催化剂,且共轭微孔聚合物/钯镍双金属催化剂为负载型手性共轭微孔聚合物/钯镍双金属纳米粒子催化剂;
所述制备方法包括如下步骤:
s1.制备含有酰胺键的手性共轭微孔聚合物;
s2.将聚合物用玛瑙研钵研磨至细小的粉末状后加入二颈瓶中,然后向二颈瓶内加入乙二醇,并对乙二醇和聚合物进行超声分散,制得载体溶液a;其中:聚合物与乙二醇的混合质量比为15~25:1;
s3.按1~1.3:1~1.5:1:1~2的质量比混合pd(oac)2、ni(oac)2·4h2o、氯仿与去离子水,搅拌溶解,制得试剂溶液b;
s4.将步骤s3制得的试剂溶液b注入步骤s2制得的载体溶液a中,室温搅拌5~7小时后,获得溶液c;
s5.向溶液c中加入硼氢化钠水溶液,搅拌5~7小时,调节ph值为中性,获得混合液d;其中:溶液c与硼氢化钠水溶液的混合质量比为40~45:1;
s6.将步骤s5制得的混合液d过滤洗涤,并真空干燥,制得负载型手性共轭微孔聚合物/钯镍双金属纳米粒子催化剂。
优选的,所述步骤s2中,聚合物与乙二醇的混合质量比为20:1。
优选的,所述步骤s4及步骤s5中,搅拌时间均为6小时,且步骤s5中,溶液c与硼氢化钠水溶液的混合质量比为43:1。
综上,所述步骤s1中,制备含有酰胺键的手性共轭微孔聚合物时,包括:
s11.在氮气保护状态下,将二氯亚砜注入装有3,5-二溴苯甲酸的三颈瓶中,混合并加热到70~80℃,在70~80℃下搅拌反应4小时后,经旋转蒸发得到化合物3,5-二溴苯甲酰氯;
s12.在氮气保护状态下,将(s)-(-)-α-苯乙胺注入装有3,5-二溴苯甲酰氯的三颈瓶中,混合后,使用微波反应器在功率为100w的条件下持续加热5~10分钟,然后以三氯甲烷为良溶剂,正己烷为不良溶剂,在1~5℃的条件下缓慢结晶,得到白色针状晶体a;
s13.将1,3,5-三溴苯,pd(pph3)2cl2,cui和pph3加入三颈烧瓶中,进行氮气保护,然后向三颈烧瓶中加入三乙胺作为溶剂,反应15分钟后加入三甲基硅乙炔,在85~95℃的条件下搅拌反应15~17小时,反应后的溶液经硅藻土抽滤,抽滤滤液经过旋转蒸发后获得粗产物b;
s14.对粗产物b进行硅胶色谱柱提纯,提纯后经旋转蒸发得到淡黄色浓溶液c,浓溶液c冷却后析出晶体化合物1,3,5-三(三甲基硅乙炔基)苯;其中:提纯时,提纯淋洗剂为正己烷,且正己烷的rf值为0.42;
s15.将1,3,5-三(三甲基硅乙炔基)苯和k2co3加入三颈瓶中,进行氮气保护,保护后向三颈烧瓶中加入四氢呋喃和甲醇,室温搅拌反应6小时,然后经过滤及旋转蒸发,除掉四氢呋喃和甲醇溶剂,获得粗产物d;
s16.对粗产物d经硅胶色谱柱进行提纯,然后旋转蒸发得到白色固体e;其中:提纯时,提纯淋洗剂为正己烷,且正己烷的rf值为0.56;
s17.将步骤s12所得的晶体a、步骤s16所得的固体e加入三颈瓶中,然后依次加入pd(pph3)2cl2、cui、pph3,接着在氮气保护下,注入三乙胺和二甲基甲酰胺,获得混合物f;
s18.将装有混合物f的三颈瓶置于微波反应器中,在功率为200w的条件下加热搅拌10~15分钟,在功率为300w的条件下加热搅拌4~6分钟,得到粗产物g;
s19.分别使用甲醇、ki水溶液、三氯甲烷对粗产物g进行洗涤,洗涤后经后干燥得到黄色固体,即为含有酰胺键的手性共轭微孔聚合物。
优选的,所述步骤s11中,二氯亚砜与3,5-二溴苯甲酸的混合质量比为4~5:1,且加热方式为油浴加热,加热温度为75℃,搅拌反应时间为4小时。
优选的,所述步骤s12中,(s)-(-)-α-苯乙胺与3,5-二溴苯甲酰氯的混合质量比为1:2~2.5,且使用微波反应器持续加热时,持续加热为8分钟,结晶温度为2℃,结晶时间为12小时。
优选的,所述步骤s13中,1,3,5-三溴苯、pd(pph3)2cl2、cui、pph3、三乙胺与三甲基硅乙炔的混合质量比为1:65~70:15~20:25~26:35~36:1.5~2,且混合后在90℃条件下搅拌反应16小时。
优选的,所述步骤s15中,1,3,5-三(三甲基硅乙炔基)苯、k2co3、四氢呋喃与甲醇的混合质量比为1:55~60:12~15:4~5。
优选的,所述步骤s17中,晶体a、固体e、pd(pph3)2cl2、cui、pph3、三乙胺与二甲基甲酰胺的混合质量比为11~12:7~8:100:2~3:6~7:1:1~1.5。
优选的,所述步骤s18中,在功率为200w的条件下加热搅拌时间为12分钟,在功率为300w的条件下加热搅拌时间为5分钟。
本发明与现有技术相比,具有以下有益效果:
在本发明所提供的制备方法中,以含有酰胺键的手性共轭微孔聚合物为载体,以乙二醇为溶剂,进行分散混合,制得载体溶液;以混合的pd(oac)2、ni(oac)2·4h2o、氯仿和去离子水为试剂溶液;混合载体溶液与试剂溶液,并在硼氢化钠还原的条件下,制得钯镍双金属纳米粒子,且钯镍双金属纳米粒子负载于手性共轭微孔聚合物上,由此获得负载型手性共轭微孔聚合物/钯镍双金属纳米粒子催化剂;上述整体反应体系在室温水浴中进行,无需高温煅烧,从而有效解决了传统浸渍法中普遍存在的金属纳米粒子稳定性差、颗粒尺寸分布不均匀、易发生团聚和能耗大等问题;
另外,基于本方法所制备的催化剂还能有效引入镍金属纳米粒子,对于提高催化活性、减少钯的投入量、降低成本具有重要意义;并且还具有稳定性好、可回收性高的优点。
附图说明
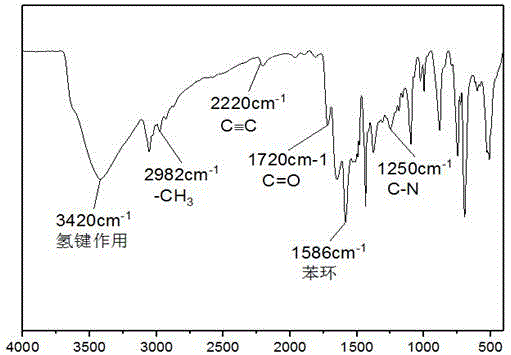
图1为通过本发明所提供的方法制得的含酰胺键手性共轭微孔聚合物的红外谱图;
图2为通过本发明所提供的方法制得的含酰胺键手性共轭微孔聚合物的扫描电镜图;
图3为通过本发明所提供的方法制得的负载型手性共轭微孔聚合物/钯镍双金属纳米粒子催化剂的x光电子能谱图;
图4为通过本发明所提供的方法制得的负载型手性共轭微孔聚合物/钯镍双金属纳米粒子催化剂的扫描透射电镜图及高角度环形暗场像图。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
在本发明中提供了一种共轭微孔聚合物/钯镍双金属催化剂的制备方法,以粉末状含有酰胺键的手性共轭微孔聚合物和乙二醇为原料,超声分散,制得溶液a;以pd(oac)2、ni(oac)2·4h2o、氯仿和去离子水为原料,搅拌混合,制得溶液b;将溶液a与溶液b混合,并通过硼氢化钠还原及室温水浴反应制得共轭微孔聚合物/钯镍双金属催化剂,且共轭微孔聚合物/钯镍双金属催化剂为负载型手性共轭微孔聚合物/钯镍双金属纳米粒子催化剂;制备方法包括如下步骤:
s1.制备含有酰胺键的手性共轭微孔聚合物;
s11.在氮气保护状态下,将二氯亚砜注入装有3,5-二溴苯甲酸的三颈瓶中,混合并加热到70~80℃,在70~80℃下搅拌反应4小时后,经旋转蒸发得到化合物3,5-二溴苯甲酰氯;
在本步骤中,二氯亚砜与3,5-二溴苯甲酸的混合质量比为4~5:1;且加热方式优选为油浴加热,加热温度可优选为75℃,搅拌反应时间可优选为4小时。
s12.在氮气保护状态下,将(s)-(-)-α-苯乙胺注入装有3,5-二溴苯甲酰氯的三颈瓶中,混合后,使用微波反应器在功率为100w的条件下持续加热5~10分钟,然后以三氯甲烷为良溶剂,正己烷为不良溶剂,在1~5℃的条件下缓慢结晶,得到白色针状晶体a;
在本步骤中,(s)-(-)-α-苯乙胺与3,5-二溴苯甲酰氯的混合质量比为1:2~2.5,且使用微波反应器持续加热时,持续加热时间可优选为8分钟,结晶温度可优选为2℃,结晶时间可优选为12小时。
s13.将1,3,5-三溴苯,pd(pph3)2cl2,cui和pph3加入三颈烧瓶中,进行氮气保护,然后向三颈烧瓶中加入三乙胺作为溶剂,反应15分钟后加入三甲基硅乙炔,在85~95℃的条件下搅拌反应15~17小时,反应后的溶液经硅藻土抽滤,抽滤滤液经过旋转蒸发后获得粗产物b;
在本步骤中,1,3,5-三溴苯、pd(pph3)2cl2、cui、pph3、三乙胺与三甲基硅乙炔的混合质量比可优选为1:65~70:15~20:25~26:35~36:1.5~2,进一步的还可优选为1:68:18:25.8:35.2:1.8,且混合后在90℃条件下搅拌反应16小时。
s14.对粗产物b进行硅胶色谱柱提纯,提纯后经旋转蒸发得到淡黄色浓溶液c,浓溶液c冷却后析出晶体化合物1,3,5-三(三甲基硅乙炔基)苯;其中:提纯时,提纯淋洗剂为正己烷,且正己烷的rf值为0.42。
s15.将1,3,5-三(三甲基硅乙炔基)苯和k2co3加入三颈瓶中,进行氮气保护,保护后向三颈烧瓶中加入四氢呋喃和甲醇,室温搅拌反应6小时,然后经过滤及旋转蒸发,除掉四氢呋喃和甲醇溶剂,获得粗产物d;
在本步骤中,1,3,5-三(三甲基硅乙炔基)苯、k2co3、四氢呋喃与甲醇的混合质量比可优选为1:55~60:12~15:4~5,进一步的还可优选为1:57:14.4:4.3。
s16.对粗产物d经硅胶色谱柱进行提纯,然后旋转蒸发得到白色固体e;其中:提纯时,提纯淋洗剂为正己烷,且正己烷的rf值为0.5。
s17.将步骤s12所得的晶体a、步骤s16所得的固体e加入三颈瓶中,然后依次加入pd(pph3)2cl2、cui、pph3,接着在氮气保护下,注入三乙胺和二甲基甲酰胺,获得混合物f;
在本步骤中,晶体a、固体e、pd(pph3)2cl2、cui、pph3、三乙胺与二甲基甲酰胺的混合质量比可优选为11~12:7~8:100:2~3:6~7:1:1~1.5,进一步的还可优选为11.3:7.1:100:2.7:6.3:1:1.4。
s18.将装有混合物f的三颈瓶置于微波反应器中,在功率为200w的条件下加热搅拌10~15分钟,在功率为300w的条件下加热搅拌4~6分钟,得到粗产物g;
在本步骤中,优选的,在功率为200w的条件下加热搅拌时间为12分钟,在功率为300w的条件下加热搅拌时间为5分钟。
s19.分别使用甲醇、ki水溶液、三氯甲烷对粗产物g进行洗涤,洗涤后经后干燥得到黄色固体,即为含有酰胺键的手性共轭微孔聚合物
s2.将聚合物用玛瑙研钵研磨至细小的粉末状后加入二颈瓶中,然后向二颈瓶内加入乙二醇,并对乙二醇和聚合物进行超声分散,制得载体溶液a;其中:聚合物与乙二醇的混合质量比为15~25:1;
在本步骤中,聚合物与乙二醇的混合质量比可优选为20:1。
s3.按1~1.3:1~1.5:1:1~2的质量比混合pd(oac)2、ni(oac)2·4h2o、氯仿与去离子水,搅拌溶解,制得试剂溶液b;
在本步骤中,关于pd(oac)2、ni(oac)2·4h2o、氯仿与去离子水的混合质量比,可进一步的优选为1:1.2:1:1.6。
s4.步骤s3制得的试剂溶液b注入步骤s2制得的载体溶液a中,室温搅拌5~7小时后,获得溶液c。
s5.向溶液c中加入硼氢化钠水溶液,搅拌5~7小时,调节ph值为中性,获得混合液d;其中:溶液c与硼氢化钠水溶液的混合质量比为40~45:1。
综上,在步骤s4及步骤s5中,搅拌时间均为6小时,且步骤s5中,溶液c与硼氢化钠水溶液的混合质量比可优选为43:1。
s6.将步骤s5制得的混合液d过滤洗涤,并真空干燥,制得负载型手性共轭微孔聚合物/钯镍双金属纳米粒子催化剂。
综上,针对上述制备方法,基于进一步的优选反应数据,在本实施例中提供如下具体制备示例:
(1)在氮气保护状态下,将二氯亚砜(5.00ml)注入装有3,5-二溴苯甲酸(1.19g,6.80mmol)的三颈瓶中,通过油浴加热到75℃搅拌反应4小时,然后经旋转蒸发得到化合物3,5-二溴苯甲酰氯;
(2)在氮气保护状态下,将(s)-(-)-α-苯乙胺(0.880ml,6.80mmol)注入装有3,5-二溴苯甲酰氯(2.03g,6.80mmol)的三颈瓶中,混合后,使用微波反应器在功率为100w的条件下持续加热8分钟,然后以三氯甲烷为良溶剂,正己烷为不良溶剂,在2℃的条件下缓慢结晶,得到白色针状晶体a;
(3)将1,3,5-三溴苯(5.00g,15.9mmol),pd(pph3)2cl2(340mg,0.490mmol),cui(90.0mg,0.490mmol)和pph3(129mg,0.490mmol)加入三颈烧瓶中,进行氮气保护,然后向三颈烧瓶中加入176ml三乙胺作为溶剂,反应15分钟后加入9.00ml三甲基硅乙炔,在90℃的条件下搅拌反应16小时,反应后的溶液经硅藻土抽滤,抽滤滤液经过旋转蒸发后获得粗产物b;获得粗产物b后,对粗产物b进行硅胶色谱柱提纯,提纯后经旋转蒸发得到淡黄色浓溶液c,浓溶液c冷却后析出晶体化合物1,3,5-三(三甲基硅乙炔基)苯;其中:提纯时,提纯淋洗剂为正己烷,且正己烷的rf值为0.42;
(4)将1,3,5-三(三甲基硅乙炔基)苯(1.00g,2.73mmol)和k2co3(57.0mg,0.410mmol)加入三颈瓶中,进行氮气保护,保护后向三颈烧瓶中加入14.4ml四氢呋喃和4.30ml甲醇,室温搅拌反应6小时,然后经过滤及旋转蒸发,除掉四氢呋喃和甲醇溶剂,获得粗产物d;获得粗产物d后,对粗产物d进行硅胶色谱柱提纯,然后旋转蒸发得到白色固体e;其中:提纯时,提纯淋洗剂为正己烷,且正己烷的rf值为0.56;
(5)将(2)中所得的晶体a(79.0mg,223μmol)、(4)中所得的固体e(57.0mg,0.410mmol)加入三颈瓶中,然后依次加入pd(pph3)2cl2(700g,100μmol)、cui(19.0mg,100μmol)、pph3(44.0mg,170μmol),接着在氮气保护下,注入7ml三乙胺和10ml二甲基甲酰胺,获得混合物f;将装有混合物f的三颈瓶置于微波反应器中,在功率为200w的条件下加热搅拌12分钟,在功率为300w的条件下加热搅拌5分钟,得到粗产物g;
(6)分别使用甲醇、ki水溶液、三氯甲烷对粗产物g进行洗涤,洗涤后经后干燥得到黄色固体,即为含有酰胺键的手性共轭微孔聚合物;
(7)将(6)中所得的聚合物(100mg)用玛瑙研钵研磨至细小的粉末状后加入二颈瓶中,然后向二颈瓶内加入乙二醇(5ml),并对乙二醇和聚合物进行超声分散,制得载体溶液a;
(8)将pd(oac)2(5.27mg,0.0230mmol)、ni(oac)2·4h2o(5.78mg,0.0230mol)加入氯仿(5ml)与去离子水(8ml)的混合液中,搅拌溶解,制得试剂溶液b;
(9)将(8)中制得的试剂溶液b注入(7)中制得的载体溶液a中,室温搅拌6小时后,获得溶液c;向溶液c中加入1m硼氢化钠水溶液(3ml),搅拌6小时,调节ph值为中性,获得混合液d;
(10)将(9)中制得的混合液d过滤洗涤,并真空干燥,制得负载型手性共轭微孔聚合物/钯镍双金属纳米粒子催化剂。
以上述具体制备示例的制备过程为基础,对其进行红外扫描、电镜扫描、x光扫描和透射扫描;具体获得:
图1所示的含酰胺键手性共轭微孔聚合物的红外谱图;
结合图1所示可知,3420cm-1处出现氢键作用的特征峰,2982cm-1处出现ch3的伸缩振动峰;2220cm-1处出现c≡c的伸缩振动峰,1720cm-1处出现酰胺中c=o的伸缩振动峰,1586cm-1处出现苯环的伸缩振动峰,1250cm-1处出现酰胺中c-n的伸缩振动峰,因此可以确定含酰胺键手性共轭微孔聚合物已成功合成。
图2所示的含酰胺键手性共轭微孔聚合物的扫描电镜图;
结合图2所示可知,所制备的聚合物尺寸为100nm左右,其形貌为纳米颗粒,具有较大的比表面积。
图3所示的含酰胺键手性共轭微孔聚合物负载钯镍纳米粒子时的x光电子能谱图(即负载型手性共轭微孔聚合物/钯镍双金属纳米粒子催化剂的x光电子能谱图);
由图3可知,由xps测得钯负载量为1.77wt%,并且从图3a中可看出pd3d的xps谱图,在335.4ev峰、340.7ev峰处分别为该催化剂的pd03d5/2和pd03d3/2,由此可知钯的价态为0价。从图3b中可看出,存在两种价态的镍,在853.5ev峰、870.4ev峰处分别为0价镍的ni2p3/2和ni2p1/2;在859.3ev峰与877.3ev峰处分别为更高氧化态的镍的nia2p3/2和nia2p1/2,这有效证明钯镍纳米粒子成功负载到了含酰胺键手性共轭微孔聚合物中。
图4所示的含酰胺键手性共轭微孔聚合物负载钯镍纳米粒子时的扫描透射电镜图和高角度环形暗场像图(即负载型手性共轭微孔聚合物/钯镍双金属纳米粒子催化剂的扫描透射电镜图和高角度环形暗场像图)。
图中4a图表示为扫描透射电镜显示的钯纳米粒子处于结晶相的图像,由图可知,晶面间距为0.224nm,由此能对应于钯的fcc(111)晶面;
另外,图中由图4b和图4c可知,高角度环形暗场像图中较亮的部分为原子序数较大的钯原子和镍原子,且能有效观察到钯原子和镍原子均匀的分散在手性共轭微孔聚合物载体上,由此进一步证实了钯镍纳米粒子成功负载到了含酰胺键手性共轭微孔聚合物中。
综上,即表明了通过本方法能有效完成负载型手性共轭微孔聚合物/钯镍双金属纳米粒子催化剂的制备。
需要说明的是,在本文中,诸如第一和第二等之类的关系术语仅仅用来将一个实体或者操作与另一个实体或操作区分开来,而不一定要求或者暗示这些实体或操作之间存在任何这种实际的关系或者顺序。而且,术语“包括”、“包含”或者其任何其他变体意在涵盖非排他性的包含,从而使得包括一系列要素的过程、方法、物品或者设备不仅包括那些要素,而且还包括没有明确列出的其他要素,或者是还包括为这种过程、方法、物品或者设备所固有的要素。
尽管已经示出和描述了本发明的实施例,对于本领域的普通技术人员而言,可以理解在不脱离本发明的原理和精神的情况下可以对这些实施例进行多种变化、修改、替换和变型,本发明的范围由所附权利要求及其等同物限定。
- 还没有人留言评论。精彩留言会获得点赞!