一种新型两性手性选择剂CEC整体柱的制备及用途的制作方法
本发明涉及手性毛细管电色谱(cec)整体柱的制备及应用技术领域,具体涉及一种双[-6-n-(咪唑基-2-氨基-丙酸-3-)]-β-cd(β-cd-e2)手性cec柱的制备及应用,即将β-cd-e2作为两性手性选择剂制备成cec整体柱的方法及对手性物质对映异构体的拆分研究和应用。
背景技术:
手性化合物在药物、环境、临床、生物和食品分析中起着重要的作用。手性农药占常用农药的40%以上,在200多种临床使用的药物中,有114种为手性药物,可见手性药物运用之广泛。植物和生物中的许多分子具有特定的对映体特性,在药理学、药代动力学、代谢和毒性等方面存在差异,其对非靶标生物或人类会造成不可预测的对映选择性生态风险。正如z.c.rao等学者2019年在nature上所阐述的:由于手性污染物的对映选择性生物降解,包括对映体结合、新陈代谢、生物清除等特定的生物学过程。对人类的健康和环境而言,生物分子区分对映异构体的能力对生命系统至关重要。因此,对映体的分离分析对于确保药物安全和环境保护具有重要意义,这一需求对微量手性分离分析领域提出了更高更迫切的要求。
β-环糊精类(β-cds)由于其优良的超分子特性已成为使用最广泛的手性选择剂之一。目前,根据β-cd衍生化官能团的不同可分为阴离子型、中性和阳离子型三种。阴离子型β-cd衍生物在分离酸性、中性和碱性手性化合物方面都具有广泛的应用。硫酸化、乙酰化、磺酰基化、磷酸化、和羧甲基化等阴离子型β-cd由于其结构中的酸性基团与手性分子亲和作用较强,由于静电相互作用易于形成包含,显现出较好的对映体选择性;作为手性选择剂的中性β-cd主要是羟丙基-β-环糊精(hp-β-cd)和天然β-cd等,中性β-cd存在情况下,疏水相互作用被认为在复杂的配合物形成过程中起主要作用;关于阳离子型β-cd作为手性选择剂用于对映体分离的研究相对阴离子型β-cd少,部分含有苯基、咪唑、铵和吡啶功能化的阳离子β-cd衍生物由于具有了各种相互作用位点,包括π-π共轭、偶极-偶极、离子对位、静电吸引和空间排斥等,对手性化合物的分离表现出巨大的潜力,烷基链较短表现出更强的手性识别能力。
毛细管电色谱(cec)是毛细管电泳和高效液相色谱的结合,是一种非常有用的混合微分离技术,采用eof(电渗流)作为驱动力;加压毛细管电色谱(pcec)将压力流引入cec系统中,有助于解决了气泡、干柱等问题,但操作较繁琐。cec对映体分离的关键要素是手性色谱柱,可以使用已经制备成功的hplc固定相,或是将固定相与其他具有足够孔隙度和功能性的材料结合,例如金纳米颗粒(gnp),二氧化硅颗粒(sinp),磁性纳米颗粒(mnp),碳纳米管和金属涂层有机框架等,更加快速准确地调整对复杂样品的对映体选择性,得以有效分离外消旋手性化合物。cds类固定相修饰的cec在对映体分离领域得到了广泛应用,包括临床药物、氨基酸、农药等手性化合物的分离。
cec柱可分为填充毛细管柱、开放管状柱和整体毛细管柱三种。与传统的cec填充柱相比,毛细管整体柱的主要优点是传质速度快,渗透率高,孔径可改变。同时,整体柱表现出令人满意的选择性和承载能力优于开管柱。到目前为止,功能化β-cd衍生物主要通过化学键合固定在整体材料上,包括三唑键、氨基键或乙醚键等共价键结合。据报道,大约有三种不同的制备方法可将β-cds键合在毛细管内,可将其表示为两步法,单步法和一锅法。
两步法首先通过原位共聚或溶胶-凝胶法制备具有适当孔结构和活性官能团的基体材料,然后通过这些活性基团与柱内壁的化学反应来固定手性选择剂,这种多步法可以有效地将柱形态与对映选择性的控制分离开来,是共价键合手性选择剂通用的方法。两步法制备整体柱,能够独立控制柱形貌,在仔细优化基质柱的形貌后,可以实现对对映选择性的调控。然而,如何以适当的耦合方式固定足够数量的手性选择剂、后修饰方法耗时长以及重复性差等问题仍然是这种制备方法的一个主要挑战。
单步法利用商业上可用的或实验室合成的手性前驱体(单体或硅烷氧化物),通过原位共聚或溶胶-凝胶法可以很容易地制备出整体柱。对于有机聚合物或有机-二氧化硅杂化csp,所需的手性选择剂需要转化为可聚合的手性单体,如甲基丙烯酸、丙烯酸或烯丙基化手性选择剂。单步法结合了整体形貌的控制和对映选择性的操控。然而,在每种情况下,为了获得满意的形貌和对映选择性,对共聚条件的选择性优化是不可避免的。由于所需的手性前驱体大多不是商品化的,它们的合成有时是困难的。此外,手性选择剂是否被封装在整体骨架内继而影响对映选择性,仍然是一个问题。
与两步法和单步法相比,一锅法是一种提高化学反应效率的策略,即反应物在单个反应容器中进行连续的化学反应,这种方法避免了中间体的冗长纯化程序。一锅法制备毛细管整体柱,因其制备工艺简单、整体柱具有更高的稳定性和更长的使用寿命而受到广泛应用。
近年来,采用β-cds作为流动相添加剂来分离手性农药的研究有见报道,但利用cds固定相修饰cec分离手性农药的研究报道较少,已成为一项具有挑战的工作。
技术实现要素:
手性选择剂的独有选择性也往往是其局限性,哪怕是商品柱分离手性化合物的能力也往往是特定的、有局限性的。针对现有技术中存在的不足,本发明的目的在于提供了一种新型两性手性高效毛细管电泳(hpce)整体柱的制备及应用技术,具体涉及了一种双[-6-氮-(咪唑基-2-氨基-丙酸-3-)]-β-环糊精(β-cd-e2)手性cec整体柱的制备及应用,即涉及一种将β-cd-e2制备成cec手性整体柱的方法及对手性化合物对映异构体的拆分应用研究。这些工作是基于以下步骤进行:(1)、用三步法合成了双[-6-氮-(咪唑基-2-氨基-丙酸-3-)]-β-环糊精(β-cd-e2)。(2)、用β-cd-e2构建手性环境,制备成手性cec整体柱,应用于高效毛细管电泳(hpce)中。(3)用所制得的β-cd-e2手性柱可建立碱性物质盐酸特拉唑嗪、盐酸异丙嗪、羟丙哌嗪和两性物质甲磺酸多沙唑嗪等手性受体拮抗剂的对映体拆分和定量分析方法,部分手性药物给出了可通过该手性柱制备获得电泳纯的单一对映体的可能性。(4)用所制得的β-cd-e2手性cec柱可拆分酸性物质咪唑乙烟酸和甲咪唑烟酸两种除草剂对映体,并建立定性定量分析方法,为获得电泳纯单一对映体提供可能。
本发明将双[-6-氮-(咪唑基-2-氨基-丙酸-3-)]-β-环糊精用于高效毛细管电泳(hpce)领域,将其制备成手性cec整体柱,用于多种手性对映体的拆分。首先将准备作为cec手性单体的β-cd-e2合成后用紫外分光光度法(uv)、傅里叶红外光谱(ftir)、元素分析、核磁共振(nmr)、液-质联用(lc-ms)等手段进行表征,确定是所要目标化合物。然后,对得到的β-cd-e2手性cec整体柱,用扫描电镜(sem)对其筛选、表征和确认,同时利用光学显微镜和扫描电镜证实了β-cd-e2整体柱内的微孔结构。该手性cec整体柱可用于盐酸特拉唑嗪(ter)、盐酸异丙嗪(pmz)、甲磺酸多沙唑嗪(dox)、羟丙哌嗪(drop)、咪唑乙烟酸(imzr)和甲咪唑烟酸(imzc)的手性拆分,得到了这4种受体拮抗剂和2种除草剂的最佳hpce分离条件,建立了相关单一对映体的电泳纯级制备和定性定量分析方法。在一根柱上同时实现了酸性、碱性及两性手性物质的理想拆分效果:本发明利用双[-6-氮-(咪唑基-2-氨基-丙酸-3-)]-β-环糊精制成的手性cec固定相,可建立多种手性物质hpce拆分及定性定量检测新方法。结果显示此种制备hpce柱的方法是有效的,可成功地将β-cd-e2作为手性单体采用一锅法键合到该cec柱内,也进一步表明此种cec柱制备法是可行的。用上述两性β-环糊精衍生物制备手性cec柱的方法是有创新性的工作,在hpce手性固定相中运用的相关工作也是有创新性的。
与现有技术相比,本发明的优点和有益效果在于:
1、申请人通过三步反应合成了β-cds:双[-6-氮-(咪唑基-2-氨基-丙酸-3-)]-β-环糊精(β-cd-e2),首次将其作为两性手性选择剂通过“一锅法”策略制备了cec整体柱,并成功应用于酸性手性农药除草剂和受体拮抗剂类碱性药物的对映体分离领域。结果表明,β-cd-e2整体柱对酸性和碱性化合物均表现出较高的对映体选择性。该方法具有快速、简单、成本低等优点。此项工作为酸碱两性手性化合物建立了一种新的cec拆分体系,同时也证明了β-cd-e2作为一种新的两性手性选择剂在手性分离分析方面具有巨大潜力。
2、首次将双[-6-氮-(咪唑基-2-氨基-丙酸-3-)]-β-环糊精作为两性手性选择剂用于进行手性拆分,未见文献报道,该技术弥补了这一领域的空白。
3、提供了一种新型β-环糊精衍生物的cec手性整体柱,确定了用双[-6-氮-(咪唑基-2-氨基-丙酸-3-)]-β-环糊精作为手性选择剂,制备手性hpce整体柱的方法,拓展了这一物质的应用范围。
4、用双[-6-氮-(咪唑基-2-氨基-丙酸-3-)]-β-环糊精衍生物制备成的手性cec整体柱,可为从消旋体中得到电泳纯的imzr、ter、dox、drop等单一对映体纯品,提供一种新方法。
5、在双[-6-氮-(咪唑基-2-氨基-丙酸-3-)]-β-环糊精衍生物手性cec整体柱上获得了可分离ter、pmz、dox、drop、imzr和imzc等手性物质的分离条件,在最佳条件下均可实现基线分离,据此可建立多种手性物质单一对映体hpce定性定量测定新方法。
附图说明
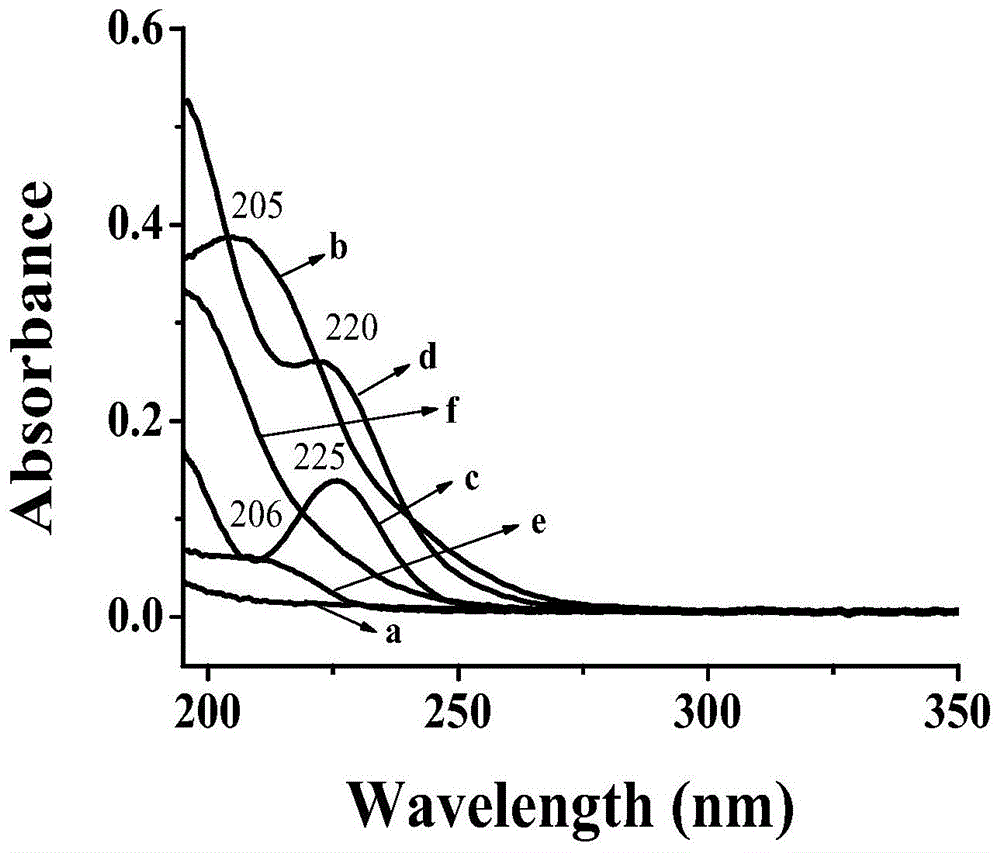
图1:紫外扫描(uv)图谱;
其中,(a)β-cd;(b)β-cd-a2;(c)ki;(d)β-cd-i2;(e)l-组氨酸;(f)β-cd-e2;
图2:傅里叶红外扫描(ftir)图谱;
其中,(a)β-cd;(b)β-cd-a2;(c)β-cd-e2;
图3:光电子xps能谱图;
其中,(a)β-cd-e2能谱图;(b)碳元素能谱图;(c)氧元素能谱图;(d)氮元素能谱图;
图4:葡聚糖环的碳位置图;
其中,(a)β-cd;(b)β-cd-a2;(c)β-cd-e2;图5:核磁共振谱图;
其中,1hnmr:(a)β-cd;(b)β-cd-a2;(c)β-cd-e2;13cnmr:(d)β-cd;(e)β-cd-a2;(f)β-cd-e2;
图6:液质联用谱图;
其中,(a)、(b)为一级质谱图;(c)、(d)、(e)为二级质谱图;
图7:质谱图中出现的ad取代碎片;
图8:质谱图中未出现的ab、ac取代碎片;
图9:β-cd-e2结构式;
图10:β-cd-e2整体柱的制备路径;
图11:光学显微镜图;
其中,(a)空柱;(b)β-cd-e2整体柱;
图12:扫描电镜图;
其中,(a)空柱扫描图(放大1250倍);(b)β-cd-e2整体柱扫描图(放大1250倍);(c)β-cd-e2整体柱扫描图(放大5000倍);(d)β-cd-e2整体柱扫描图(放大10000倍);
图13:盐酸特拉唑嗪消旋体拆分电泳图;
图14:盐酸异丙嗪分离电泳图;
图15:羟丙哌嗪分离电泳图;
图16:磺酸多沙唑嗪分离电泳图;
图17:咪唑乙烟酸拆分对照实验图;
其中,(a)溶剂(h2o);(b)空柱分离咪唑乙烟酸;(c)β-cd-e2整体柱拆分咪唑乙烟酸;
图18:甲咪唑烟酸拆分电泳图。
具体实施方式
下面申请人将结合具体的实施例对本发明的新手性cec整体柱的制备和应用过程做详细说明,目的在于使本领域技术人员对本发明有更进一步的理解。
实施例1:双[-6-氮-(咪唑基-2-氨基-丙酸-3-)]-β-cd的制备与表征
依据文献(宋发军,丁志刚等.β-环糊精衍生物的金属配合物催化rna的水解.分子催化,2001,15(02):139-142.)用三步法制得β-cd-e2后,采用紫外分光光度法(uv)、傅里叶红外光谱(ftir)、元素分析、核磁共振(nmr)、液-质联用(lc-ms)等手段对β-cd-e2单体进行了表征,确定是所要目标化合物。
具体做法:先将顺丁烯二酸酐和β-cd(摩尔比15:1)两者反应,后处理,烘干产物β-cd-a2,备用;为了在β-cd侧链上引入活泼的取代基团,按上述文献将合成的β-cd-a2与ki反应后的产物β-cd-i2后处理提纯,抽滤、干燥备用;接着,按上述文献将合成的β-cd-i2与组氨酸反应得到目标产物β-cd-e2,经后处理提纯后,干燥备用。
β-cd-e2的合成方法具体反应过程如下所示:
1、紫外扫描(uv)图谱
图1中的所有化合物均以超纯水为溶剂,浓度为1.0×10-5mol/l。由图1可知,β-cd结构中无刚性结构,存在c-c、c-o和-oh化学键和n-σ*、σ-σ*跃迁,在200~600nm范围无强紫外吸收;β-cd-a2上马来酸酐基团的引入,使得结构中存在c=c、c=o和π-π共轭效应,所以在205nm左右处有强吸收;i-由于极大地削弱水分子之间氢键,在225nm处有强吸收,β-cd-i2在220nm处出现强吸收;β-cd-e2由于羧基的吸电子效应使得组氨酸的特征吸收峰(210nm)发生蓝移,在200nm处出现强吸收,可初步说明结构上引入了组氨酸基团。
2、傅里叶红外扫描(ftir)图谱
图2中结果显示,由于天然β-cd分子空腔外含有众多-oh官能团,红外谱图中出现了3380cm-1的-oh的伸缩振动峰(3600~3200cm-1);β-cd-a2的红外谱图中存在1730cm-1处c=o(1800-1660cm-1)的伸缩振动峰和1637cm-1处c=c(1680-1610cm-1)的伸缩振动峰,表明马来酸酐已键合到β-cd上;β-cd-e2的谱图中能看到1650cm-1处c-n(1680-1640cm-1)的伸缩振动峰,以及1250cm-1和1150cm-1处c=n(1350-1100cm-1)的伸缩振动峰,进一步说明组氨酸基团的引入。
3、元素分析
将相应理论值和实测值列于表1和表2。
β-cd-a2、β-cd-i2中几乎不含n元素,β-cd-e2中n元素含量为3.82%,由此可以推测含有n元素的组氨酸基团已经键合上去。β-cd-e2中含c(绝对误差-1.21%,相对误差-2.97%)、h(绝对误差0.12%,相对误差1.91%)量测定值与理论计算值差异较小,含n量(绝对误差-1.60%,相对误差-29.52%)测定值差异略大,n元素含量低于理论计算值,这可能是由于目标产物结构中带有结晶水导致。根据元素分析结果可知,含有n元素的基团已经成功键合到β-cd上去。实际分子式基本与理论推测分子式相符:c54h84n6o37。
表1元素分析理论值
表2元素分析实测值
4、x-射线光电子能谱(xps)
对β-cd-e2进行xps能谱扫描,得到如图3所示全谱和c、o、n元素的能谱图。由图3可知,c1s的结合能为284.63ev,o1s的结合能为532.03ev,n1s的结合能为398.85ev。
5、核磁共振1h和13c(nmr)
使用400mhz*/avanceiii核磁仪对β-cd、β-cd-a2和β-cd-e2进行表征,得到1hnmr和13cnmr谱图如图5所示(c编号与h的编号一致)。以数字标示β-环糊精葡聚糖单元上c的位置,如图4所示,由核磁谱图中所得数据列于表3中。
1h和13cnmr表征均以二甲基亚砜为溶剂。由1h核磁结果分析可知,与天然β-cd相比,β-cd-a2和β-cd-e2结构中c-6位的羟基(oh-6)的化学位移4.5ppm均消失,表明马来酸酐的加成反应发生在β-cd-a2的c-6位置上;β-cd-e2较β-cd-a2新出现了2.51、2.09、2.73、2.89、3.64、5.78、6.03和7.95ppm对应组氨酸上的化学位移。由13cnmr核磁结果分析可知,β-cd-a2和β-cd-e2上c-6位的化学位移较β-cd向低场移动,由60.4ppm到60.9ppm,结合1h结果可说明组氨酸引入到了β-cd-e2的c-6位上;同时,β-cd-e2谱图中出现167.7ppm、162.8ppm、136.6ppm、124.5ppm、116.9ppm和85.4pm的化学位移,对应于c7-c12的位移。综合上述结果,表明组氨酸已键合到β-cd上,且发生取代的是在β-cd的c-6位置上。
表31h和13c谱数据解析
6、液-质联用(lc-ms)
为了进一步确定β-cd-e2葡聚糖环的取代位置,使用agilentg6520q-tof液质联用仪对β-cd-e2进行了一级、二级质谱表征,所得质谱结果如图6所示。
质谱图中存在m/z=1406.3、1266.1、1133.5、921.8、624.3、615.3、486.0的碎片离子峰。m/z=1406.3为β-cd-e2(m/z1408)母核的分子离子峰(m/z-h);m/z=1266.1为母离子断裂一分子取代碳链(c7-c12,m/z139)的产物,m/z=1133.5为断裂两分子取代碳链(m/z278)的离子峰,由此推测β-cd-e2上葡聚糖环被双取代;m/z=921.8为a、d取代的四个葡聚糖加两分子组氨酸取代基的裂解碎片,m/z=486推测为β-cd-e2分子断裂的3个葡聚糖单元的碎片,m/z=624.3和m/z=615.3分别为β-cd-e2裂解多步后得到的碎片离子峰(图7a和7b)。同时,假设β-cd上a、b葡聚糖被双取代,则β-cd-e2的裂解碎片应包含有m/z=810,598(图8a);假设β-cd上a、c葡聚糖被双取代,则β-cd-e2的裂解碎片应包含有m/z=648,760(图8b)。在获得的质谱图中均未出现该类碎片离子峰。综合上述结果,可确定在β-cd葡聚糖环的上发生的是双取代,并且组氨酸取代基的位置在7个葡聚糖环的a、d环上。
综合以上表征结果可知,β-cd结构上发生的是组氨酸双取代反应,且取代的位置在β-cd的葡聚糖单元环的a、d环上,以字母标示β-cd-e2上葡聚糖单元环的编号,得到的产物分子结构如图9所示。
实施例2双[-6-氮-(咪唑基-2-氨基-丙酸-3-)]-β-cd电色谱整体柱的制备与表征
β-cd-e2整体柱的制备路径如图10所示,采用“一锅法”按照如下步骤制备cec整体柱:
(1)在整体柱制备之前,通过甲醇、naoh溶液(0.01mol/l)、hcl溶液(0.01mol/l)、h2o依次冲洗毛细管(内径75μm,长度50cm,河北永年光导纤维厂)。然后将甲醇和kh560以1:1体积比混合后通过氮气流注入毛细管柱内;(2)将β-cd-e2(400μg)、1,4-丁二醇(129μl)、正丙醇(659μl)、乙二醇二甲基丙烯酸酯(73μl)、甲基丙烯酸缩水甘油酯(129μl)和偶氮二异丁腈(3mg)放到10ml小瓶中,将所得混合溶液填充到预处理过的毛细管柱中,有效长度为40cm,用橡胶塞密封,然后在50℃“一锅”反应5小时聚合完成后,通过氮气流冲洗毛细管以除去未反应的化学物质,冲洗完成后毛细管柱两端用硅胶封口备用。
采用光学显微镜对空柱和β-cd-e2整体柱进行显微镜成像,放大450倍,得到的结果如图11所示。
采用jsm-6700f扫描电镜对β-cd-e2手性cec整体柱进行表征,并与内壁未经任何修饰的空毛细管柱做对比,所得结果如图12所示。
由所得的sem图可以看出,a图中未经修饰的空毛细管柱内壁较光滑,孔径较大(75μm)。由图b、c、d可知,“一锅法”制备的整体柱内充满了“簇”状聚合物,形成了均匀的整体床层;由1250、5000和10000倍图像可看出,柱内既存在团簇结构,又分布着均匀的孔径,孔径大小约5μm左右,有利于流动相的流动,使得整体柱具有良好的渗透性能。结果表明,所制备的cec整体柱内形成了均质整体结构,且柱内形成了“颗粒”与“颗粒”,“簇”与“簇”的微孔结构。
实施例3用实施例2制备的双[-6-氮-(咪唑基-2-氨基-丙酸-3-)]-β-cd电色谱整体柱分离六种手性化合物
在β-cd-e2手性cec整体柱上实现了盐酸特拉唑嗪(ter)(分析标准品,消旋体,阿拉丁试剂有限公司)、盐酸异丙嗪(pmz)(分析标准品,消旋体,阿拉丁试剂有限公司)、甲磺酸多沙唑嗪(dox)(分析标准品,消旋体,阿拉丁试剂有限公司)、羟丙哌嗪(drop)(分析标准品,消旋体,阿拉丁试剂有限公司)、咪唑乙烟酸(imzr)(分析标准品,消旋体,阿拉丁试剂有限公司)和甲咪唑烟酸(imzc)(分析标准品,消旋体,阿拉丁试剂有限公司)的手性拆分,均能达到基线分离。
1、盐酸特拉唑嗪的分离
在β-cd-e2整体柱上拆分了手性药物盐酸特拉唑嗪对映体,并探究了不同缓冲体系、缓冲液ph和分离电压对其对映体分离的影响。最佳分离条件下(检测波长254nm,25℃,50mmol·l-1tris-柠檬酸缓冲体系,ph6.00,分离电压-20kv(负电源hpce仪),样品浓度2.0×10-5mol·l-1,手动进样30s),实现了盐酸特拉唑嗪两对映体的基线分离,如图13所示。本研究建立了一种盐酸特拉唑嗪手性拆分的cec新柱方法。
从图13可以看出,两对映体的保留时间分别为3.139分钟和20.108分钟,分离度rs可达41.64。可据此获得电泳纯的单一对映体。
2、盐酸异丙嗪的分离
在β-cd-e2整体柱上拆分了手性药物盐酸异丙嗪对映体,并探究了缓冲液ph、分离电压和缓冲液浓度对其对映体分离的影响。最佳分离条件下(检测波长254nm,80mmol·l-1,ph5.30的tris-磷酸缓冲液中,样品浓度量4.3×10-4mol·l-1,25℃,电压24kv,进样压力0.8psi,进样时间15s),20min内完成了盐酸异丙嗪对映体的有效分离。
分离结果如图14所示。两对映体的保留时间分别为6.604分钟和19.288分钟,分离度rs达到7.80,重复进样实验表明(n=6),盐酸异丙嗪两对映体保留时间的rsd值分别为1.08%和1.94%,两对映体峰面积的rsd值分别为7.81%和2.83%,两对映体峰高的rsd值分别为6.12%和2.60%,β-cd-e2整体柱分离盐酸异丙嗪具有良好的重现性。建立了一种盐酸异丙嗪手性拆分的cec新方法。
3、羟丙哌嗪的分离
在β-cd-e2整体柱上,30mmol·l-1na2hpo4-柠檬酸缓冲液中,ph3.50,测定波长为254nm,负电源电压为-20kv,25℃,羟丙哌嗪浓度2.0×10-5mol·l-1,压力进样20s,得到分离电泳图。
如图15所示,两对映体的保留时间分别为2.971分钟和66.441分钟,分离度rs达到158.58,可据此获得电泳纯的单一对映体,还可由此建立一种羟丙哌嗪手性拆分的cec新方法。
4、甲磺酸多沙唑嗪的分离
在β-cd-e2整体柱上,50mmol·l-1tris-磷酸缓冲液中,ph4.00,测定波长254nm,25℃,测定电压20kv,进样时间30s,甲磺酸多沙唑嗪浓度6.86×10-7mol·l-1条件下得到电泳拆分图,
如图16所示。两对映体的保留时间分别为3.407分钟和55.930分钟,分离度rs达到175.08,可据此获得电泳纯的单一对映体。还可建立一种磺酸多沙唑嗪手性拆分的cec新方法。
5、咪唑乙烟酸的分离
在β-cd-e2整体柱上拆分了手性除草剂咪唑乙烟酸对映体,并探究了缓冲液ph、分离电压和缓冲液浓度对其对映体分离的影响以及咪唑乙烟酸对映体分离的线性范围和精密度。最佳分离条件下(检测波长254nm,30mmol·l-1ph4.00的na2hpo4-柠檬酸缓冲液中,样品浓度量6.2×10-5mol·l-1,电压-20kv,手动进样20s),30min内完成了咪唑乙烟酸对映体的有效分离,分离度rs为62.65,可据此获得电泳纯的单一对映体。用实验所得最佳条件下进行空柱和溶剂对照实验。
结果如图17所示,未经修饰的空柱对咪唑乙烟酸不具手性识别能力,溶剂对响应信号无明显贡献,而用β-cd-e2制备的cec整体柱对咪唑乙烟酸对映体有明显的拆分能力。同时获得咪唑乙烟酸消旋体浓度在6.2×10-6~6.2×10-5mol·l-1的范围内,两对映体与峰面积的线性相关系数r分别为0.9973、0.9576,与峰高的线性相关系数r分别为0.9619和0.9087。最佳条件下,6次测定结果显示,咪唑乙烟酸两对映体保留时间的rsd值分别为2.07%和1.86%,两对映体峰面积的rsd值分别为7.82%和6.23%,两对映体峰高的rsd值分别为7.05%和4.67%,表明β-cd-e2整体柱分离咪唑乙烟酸具有良好的重现性。
6、甲咪唑烟酸的分离
用制备的β-cd-e2整体柱,选择检测波长254nm,在35mmol·l-1na2hpo4-柠檬酸缓冲液中,ph4.00,进样浓度2.2×10-6mol·l-1,手动进样15s的条件下得到电泳拆分图。
如图18所示,两对映体的保留时间分别为5.923分钟和7.534分钟,分离度rs达到6.35,由此可建立一种甲咪唑烟酸手性拆分的cec新方法。
- 还没有人留言评论。精彩留言会获得点赞!