一种Ce-Ni/Ca-SAPO-34催化剂及其制备方法与流程
本发明属于催化剂领域,具体涉及一种ce-ni/ca-sapo-34催化剂及其制备方法。
背景技术:
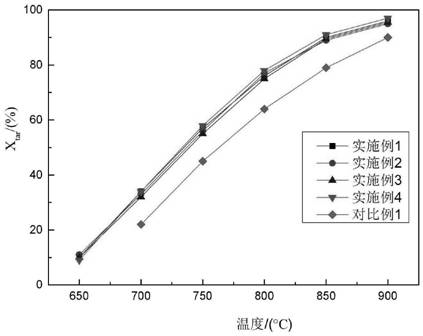
:生物质能源是所有的可再生清洁能源中唯一的碳源,它易于运输和储存。生物质热解气化技术是利用生物质能源的最有效的方法之一,可以将低品位能源转化为高品位气体和液体燃料。但热解气化过程中伴随着产生的焦油,对于仪器、产物乃至人身健康都有极大的危害。它的温度处于200℃以下时易发生冷凝,同时又容易和粉尘、焦炭等结合形成粘稠状物质,进而堵塞管道和腐蚀设备;生物质气化产生的焦油占总能量的5-10%,会降低生物质能的利用效率;焦油和其他化合物聚合,会形成更复杂的有机物,产生巨大的毒性,难以去除;焦油热解气中形成的焦油气溶胶,易被人体呼吸道吸收;焦油具有致癌性,严重影响人体的健康。焦油的严重危害,是制约生物质能源应用的重要因素之一,因此必须去除产物中的焦油。现有技术中对于焦油的去除多为热化学法,具体又分为热裂解法和催化裂解法。热裂解法是在1100℃以上的高温下,焦油化学键断裂,分子质量较大的化合物裂解形成分子质量较小的气态化合物。但是该方法的设备温度要求过高,能耗高,成本高,易生焦;在实际应用中难以达到1000-1200℃。而催化裂解法是指焦油在相对较低的温度下加入合适的催化剂,使焦油转化成小分子化合物。但是现有技术中的催化剂不能完全去除产物中的焦油,去除率在90%-95%左右;而且达到90%以上去除率所需时间长;由于在分解碳氢化合物中的重组分时表面易产生积碳,进而使催化剂失活,所以催化剂的使用寿命短。技术实现要素:为了改善现有技术中存在的生物质焦油不能从产物中完全消除、达到高去除率所需时间长和使用寿命短的问题,本发明提供一种ce-ni/ca-sapo-34催化剂及其制备方法。为实现上述目的,本发明采用以下技术方案:一种ce-ni/ca-sapo-34催化剂的制备方法,包括以下步骤:步骤一:将一定量的异丙醇铝加入适量的磷酸水溶液中以35-42℃的温度水浴搅拌2-3h,然后向溶液中滴入适量的硝酸钙水溶液,超声10-20min。随后向混合溶液中缓慢滴入硅溶胶和四乙基氢氧化铵的混合水溶液,并在85-95℃的水浴温度下快速回流搅拌3-4h获得均匀的晶化液,然后让晶化液在室温下陈化12-18h。步骤二:将陈化后的溶液放入反应釜的聚四氟乙烯内衬中以180-210℃的温度反应24-36h。反应结束后将产物离心洗涤,洗涤时用无水乙醇洗1次后用去离子水洗2次,然后放入真空冷冻干燥器中24h;冻干后的粉末在马弗炉中通过不同的升温速率升温至700℃后煅烧6-8h,冷却至室温后获得ca-sapo-34分子筛。步骤三:将六水合硝酸铈和无水二氯化镍以一定的比例溶于去离子水中,超声搅拌15-20min;然后向混合溶液中加入一定量的ca-sapo-34分子筛,并在50-60℃的水浴下连续搅拌4-6h,随后把溶液放入反应釜中以100-110℃的温度反应24-36h。然后将产物离心并以去离子水洗涤后在氮气保护下缓慢升温至650℃煅烧5-8h,获得ce-ni/ca-sapo-34催化剂。所述步骤一中异丙醇铝的添加量为0.01-0.02mol,异丙醇铝和磷酸的物质的量之比为1:1.1-1:1.5,硝酸钙和异丙醇铝的物质的量之比为1:20-1:10,异丙醇铝和硅溶胶的质量比为1:1.38-1:1.73,硅溶胶的质量分数为30wt%,异丙醇铝和四乙基氢氧化铵的物质的量之比为1:4-1:8。所述步骤三中ca-sapo-34的添加量为0.5-0.8g,ca-sapo-34和无水二氯化镍的质量比为1:0.44-1:0.77,ca-sapo-34和六水合硝酸铈的质量比为1:0.43-1:0.71。优选地,所述步骤一中磷酸水溶液的浓度为0.25-0.4g/ml;硅溶胶和四乙基氢氧化铵的混合水溶液的滴入速度为2ml/min。优选地,所述步骤一中硅溶胶和四乙基氢氧化铵的混合溶液制备时先将硅溶胶溶于20ml的去离子水中,然后将四乙基氢氧化铵缓慢的加入溶液中,并室温搅拌2-3h。优选地,所述步骤二中粉末在马弗炉中先以6℃/min的速率升温到300℃后,再以3℃/min的速度缓慢升温至700℃。优选地,所述步骤三管式炉的升温速率始终控制在2℃/min。本发明还提供另一技术方案,上述方法制备得到的ce-ni/ca-sapo-34催化剂,其中ca-sapo-34分子筛中ca:si:p:al的物质的量之比为(0.05-0.1):(0.4-0.5):(1.1-1.5):1;而ni在分子筛上的负载量为20-35wt%,ce在分子筛上的负载量为14-23wt%。有益效果:本发明提供的一种ce-ni/ca-sapo-34催化剂的制备方法制备得到的催化剂,有效改善现有技术中产物中焦油去除不彻底,高去除率所需时间长,催化剂易失活的问题。本发明通过ca-sapo-34的制备,进一步增强了对于生物质热解产物中焦油的选择性,使催化剂能够准确的催化裂解焦油,这一方面能够提高焦油的去除速度,另一方面为彻底去除产物中的焦油提供了基础。镍负载在分子筛上后由于分子筛孔洞大且密集,可以更好的催化通过分子筛的热解产物中的焦油。而铈在煅烧后形成的ceo2不仅可以改变ni的电子密度,促进ni物种的还原,从而加速碳物种的转移,并抑制碳沉积的产生;而且较高的ce含量有利于烯烃的形成,促进了氢化或脱氢,对于酚类等物质也有一定的选择性。附图说明图1为本发明所述实施例1催化剂的5μm的扫描电子显微镜图;图2为本发明所述实施例1催化剂的5nm的高分辨率的透射电镜图;图3为本发明所述实施例1-4和对比例1在催化生物质焦油时的焦油转化率xtar折线图;图4为本发明所述实施例1和对比例1在催化生物质焦油时的气体总产率yt折线图;图5为本发明所述实施例3和对比例1在催化300h后的焦油转化率与初始焦油转化率的对比柱状图。具体实施方式下面结合具体实施例对本发明进行详细说明。以下实施例将有助于本领域的技术人员进一步理解本发明,但不以任何形式限制本发明。应当指出的是,对本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进。这些都属于本发明的保护范围。实施例1步骤一:将1.08g的磷酸溶于4.3ml的去离子水中,然后向溶液中加入2.04g异丙醇铝,并以35℃的温度水浴搅拌2h,然后向溶液中滴入0.164g的硝酸钙水溶液,超声10min获得溶液a。将2.82g的硅溶胶溶于20ml的去离子水中,然后缓慢的加入11.76g的四乙基氢氧化铵,并室温搅拌3h获得溶液b。随后向溶液a中以2ml/min的速度滴入溶液b,并在85℃的水浴温度下快速回流搅拌4h获得均匀的晶化液,然后让晶化液在室温下陈化18h。步骤二:将陈化后的溶液放入反应釜的聚四氟乙烯内衬中以210℃的温度反应24h。反应结束后将产物离心洗涤,洗涤时用无水乙醇洗1次后用去离子水洗2次,然后放入真空冷冻干燥器中24h;冻干后将粉末在马弗炉中先以6℃/min的速率升温300℃,再以3℃/min的速度升温至700℃后煅烧8h,冷却至室温后获得ca-sapo-34分子筛。步骤三:将0.335g六水合硝酸铈和0.22g无水二氯化镍溶于去50ml离子水中,超声搅拌20min;然后向混合溶液中加入0.5g的ca-sapo-34分子筛,并在50℃的水浴下连续搅拌6h,随后把溶液放入反应釜中以110℃的温度反应24h。然后将产物离心并以去离子水洗涤后在氮气保护下以2℃/min升温至650℃煅烧8h,获得ce-ni/ca-sapo-34催化剂。实施例2步骤一:将3.33g的磷酸溶于8.3ml的去离子水中,然后向溶液中加入4.08g异丙醇铝,并以42℃的温度水浴搅拌3h,然后向溶液中滴入0.164g的硝酸钙水溶液,超声20min获得溶液a。将7.01g的硅溶胶溶于20ml的去离子水中,然后缓慢的加入11.76g的四乙基氢氧化铵,并室温搅拌2h获得溶液b。随后向溶液a中以2ml/min的速度滴入溶液b,并在95℃的水浴温度下快速回流搅拌3h获得均匀的晶化液,然后让晶化液在室温下陈化12h。步骤二:将陈化后的溶液放入反应釜的聚四氟乙烯内衬中以180℃的温度反应36h。反应结束后将产物离心洗涤,洗涤时用无水乙醇洗1次后用去离子水洗2次,然后放入真空冷冻干燥器中24h;冻干后将粉末在马弗炉中先以6℃/min的速率升温300℃,再以3℃/min的速度升温至700℃后煅烧6h,冷却至室温后获得ca-sapo-34分子筛。步骤三:将0.344g六水合硝酸铈和0.616g无水二氯化镍溶于去50ml离子水中,超声搅拌15min;然后向混合溶液中加入0.8g的ca-sapo-34分子筛,并在60℃的水浴下连续搅拌4h,随后把溶液放入反应釜中以100℃的温度反应36h。然后将产物离心并以去离子水洗涤后在氮气保护下以2℃/min升温至650℃煅烧5h,获得ce-ni/ca-sapo-34催化剂。实施例3步骤一:将3.0g的磷酸溶于8ml的去离子水中,然后向溶液中加入3.2g异丙醇铝,并以40℃的温度水浴搅拌2.5h,然后向溶液中滴入0.171g的硝酸钙水溶液,超声15min获得溶液a。将4.8g的硅溶胶溶于15ml的去离子水中,然后缓慢的加入10.2g的四乙基氢氧化铵,并室温搅拌2.5h获得溶液b。随后向溶液a中以2ml/min的速度滴入溶液b,并在90℃的水浴温度下快速回流搅拌3.5h获得均匀的晶化液,然后让晶化液在室温下陈化16h。步骤二:将陈化后的溶液放入反应釜的聚四氟乙烯内衬中以200℃的温度反应30h。反应结束后将产物离心洗涤,洗涤时用无水乙醇洗1次后用去离子水洗2次,然后放入真空冷冻干燥器中24h;冻干后将粉末在马弗炉中先以6℃/min的速率升温300℃,再以3℃/min的速度升温至700℃后煅烧7h,冷却至室温后获得ca-sapo-34分子筛。步骤三:将0.462g六水合硝酸铈和0.476g无水二氯化镍溶于去50ml离子水中,超声搅拌18min;然后向混合溶液中加入0.7g的ca-sapo-34分子筛,并在56℃的水浴下连续搅拌5h,随后把溶液放入反应釜中以106℃的温度反应32h。然后将产物离心并以去离子水洗涤后在氮气保护下以2℃/min升温至650℃煅烧7h,获得ce-ni/ca-sapo-34催化剂。实施例4步骤一:将1.73g的磷酸溶于5.8ml的去离子水中,然后向溶液中加入3.8g异丙醇铝,并以38℃的温度水浴搅拌2.6h,然后向溶液中滴入0.2g的硝酸钙水溶液,超声12min获得溶液a。将5.32g的硅溶胶溶于20ml的去离子水中,然后缓慢的加入13.0g的四乙基氢氧化铵,并室温搅拌2.8h获得溶液b。随后向溶液a中以2ml/min的速度滴入溶液b,并在88℃的水浴温度下快速回流搅拌3.4h获得均匀的晶化液,然后让晶化液在室温下陈化13h。步骤二:将陈化后的溶液放入反应釜的聚四氟乙烯内衬中以190℃的温度反应29h。反应结束后将产物离心洗涤,洗涤时用无水乙醇洗1次后用去离子水洗2次,然后放入真空冷冻干燥器中24h;冻干后将粉末在马弗炉中先以6℃/min的速率升温300℃,再以3℃/min的速度升温至700℃后煅烧6.5h,冷却至室温后获得ca-sapo-34分子筛。步骤三:将0.33g六水合硝酸铈和0.42g无水二氯化镍溶于去50ml离子水中,超声搅拌19min;然后向混合溶液中加入0.6g的ca-sapo-34分子筛,并在58℃的水浴下连续搅拌4.5h,随后把溶液放入反应釜中以106℃的温度反应34h。然后将产物离心并以去离子水洗涤后在氮气保护下以2℃/min升温至650℃煅烧7h,获得ce-ni/ca-sapo-34催化剂。对比例1从cn108970616a提供的制备方法制备得到现有技术的生物质焦油催化剂,具体步骤是:将硝酸镍晶体溶入蒸馏水中配成溶液,将白云石粉碎成一定粒度与硝酸镍溶液湿混,加入黏合剂和成型剂,干燥,压片成型,一定温度煅烧一段时间,粉碎过筛得到镍/白云石催化剂。具体分析如下:图1是实施例1制备的ce-ni/ca-sapo-34在5μm尺寸下的扫描电子显微镜图片,图2是5nm的高分辨率的透射电镜图片。从图1中可以看出催化剂中的ca掺杂的sapo-34在形貌上从发生了变化,成为了类长方体;而镍和铈在煅烧后的氧化物则ca-sapo-34基体的表面。从图2的高分辨透射电镜图可以看出复合催化剂的晶粒直径大概在10nm-20nm。为了验证本发明所述制备方法制备得到的催化剂在热解焦油中的作用,将实施例1-4的ce-ni/ca-sapo-34催化剂和对比例1的镍/白云石催化剂的进行焦油裂解的转化,并对焦油转化率xtar和气体总产率yt两个主要的数据进行了检测。其中焦油转化率xtar是指转化为小分子气体的焦油占进入到整个系统中的焦油的质量比例,单位为%;而气体总产率yt是指单位质量的焦油经催化裂解后产生的小分子气体产量,单位为nm3/kgtar。从图中可以看出,实施例1-4制备的ce-ni/ca-sapo-34催化剂在催化裂解生物质焦油从650℃就可以开始,并且达到相同的焦油转化率所需温度比对比例1低了40℃左右。而表1是实施例1-4和对比例1的所能达到的最高焦油转化率的统计表,从表中可以看出本发明所述制备方法制备的复合催化剂的焦油转化率都能达到98.8%以上,最高可达99.6%;而对比例1的最高焦油转化率为93.2%;通过对比可以发现本发明所述制备方法制备的复合催化剂具有十分优秀的焦油去除效果。表1实施例1-4和对比例1的焦油转化率实施例1实施例2实施例3实施例4对比例1焦油转化率98.8%99.1%99.6%99.3%93.2%从图4的实施例1和对比例1的气体总产率折线图可以发现在气体总产率方面实施例1的复合催化剂也具有更好的效果,不仅在相同温度下实施例1的气体总产率更高,而且最高的气体总产率4.5左右。这进一步证明了本发明所述制备方法制备的复合催化剂在生物质焦油的催化上相对于现有技术的进步。图5是把实施例3和对比例1连续催化300h之后的焦油转化率和最开始时的对比柱状图。非常直接的显示了实施例3和对比例1在长时间催化后的性能下降,可以看出实施例3的性能下降有限,而对比例1的性能下降幅度是实施例3的8-9倍,这说明本发明所述制备方法制备的复合催化剂具有更好的使用寿命。以上所述,仅为本发明的较佳实施例,并非对本发明任何形式上和实质上的限制,应当指出,对于本
技术领域:
的普通技术人员,在不脱离本发明方法的前提下,还将可以做出若干改进和补充,这些改进和补充也应视为本发明的保护范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1