一种非均相双金属中心金属卟啉及其制备方法和应用与流程
(一)技术领域
本发明涉及一种非均相双金属中心金属卟啉以及制备方法和其在环烷烃催化氧化中的应用,属于有机催化和精细有机合成领域。
(二)
背景技术:
环烷烃催化氧化是一个重要的化工转化过程,其氧化产物环烷醇、环烷酮不仅是重要的有机溶剂,也是重要的精细化工中间体,广泛用于农药、医药、染料等精细化工产品的合成(wo2019046316;wo2019030294;wo2019069911;cn108864082;cn109180556;journalofmedicinalchemistry2019,62:1837-1858;russianjournalofgeneralchemistry2018,88:2646-2652,即世界专利2019046316;世界专利2019030294;世界专利2019069911;中国专利108864082;中国专利109180556;药物化学杂志2019,62:1837-1858;俄罗斯基础化学杂志2018,88:2646-2652)。除此以外,环烷醇和环烷酮还可以进一步氧化制备脂肪族二酸,脂肪族二酸是制备各种高分子材料的重要前体,如环己烷催化氧化的主要产物环己醇和环己酮进一步氧化可以得到生产尼龙-66和尼龙-6的重要前体己二酸,市场需求非常大(appliedcatalysisa,general2019,575:120-131;chemicalengineeringscience2019,203:163-172;appliedcatalysisa,general2018,554:71-79;industrial&engineeringchemistryresearch2017,56:15030-15037,即基础应用催化a,2019,575:120-131;化学工程科学2019,203:163-172;基础应用催化a,2018,554:71-79;工业与工程化学研究2017,56:15030-15037)。目前,工业上环烷烃催化氧化主要是以均相钴(ii)盐或锰(ii)盐为催化剂,分子氧或空气为氧化剂,在130~160℃,0.80~3.0mpa压力下实现的(appliedcatalysisa,general2019,575:120-131;science2014,346:1495-1498,即基础应用催化a,2019,575:120-131;科学,2014,346:1495-1498)。由于反应温度高,生成的环烷醇和环烷酮易于深度氧化生成脂肪族二酸,堵塞环烷烃催化氧化过程的管道,不利于连续化生产。产生上述问题的主要根源为:氧化中间产物,环烷基过氧化氢以自由基热分解路径向目标氧化产物环烷醇和环烷酮转化,增加了反应体系的不可控性,降低了环烷基醇和环烷基酮的选择性。因此,有效控制o2催化氧化环烷烃过程中,氧化中间产物环烷基过氧化氢的催化转化,将有利于环烷烃催化氧化选择性的提高,将是工业上环烷烃催化氧化领域一项十分新颖并且应用意义极大的工艺改进,也是环烷烃催化氧化工业的迫切需求,具有极大的生产应用价值,也具有重要的理论研究价值。
金属卟啉作为细胞色素p-450的模型化合物,广泛应用于仿生催化各类有机合成反应,尤其是氧化反应(polyhedron2019,163:144-152;journalofcatalysis2019,369:133-142,即多面体2019,163:144-152;催化杂志2019,369:133-142)。金属卟啉具有近似平面的分子结构,使具有催化活性的金属中心能够最大限度地暴露在催化体系中发挥作用,在底物物质的量的1/1000000~1/100000就可以表现出优异的催化活性,能够显著降低环烷烃催化氧化的成本,是环烷烃催化氧化优选的催化剂之一。
(三)
技术实现要素:
为了克服现有技术的不足,本发明的目的在于提供一种非均相双金属中心金属卟啉及其制备方法和其在环烷烃催化氧化中的应用,以双金属中心金属卟啉为催化剂,催化o2氧化环烷烃,并有效调控氧化中间产物环烷基过氧化氢的催化转化,不仅具有环烷基醇和环烷基酮选择性高,反应温度低,副产物少,环境影响小等优势,而且本发明所提供的方法环烷基氢过氧化物含量低,安全系数高,是一种高效、可行、安全的环烷烃选择性催化氧化合成环烷基醇和环烷基酮的方法
本发明的技术方案如下:
一种非均相双金属中心金属卟啉催化剂,所述非均相双金属中心金属卟啉具有如式(i)所示结构:
式(i)中,r1、r2、r3、r4、r5各自独立为:氢、甲基、乙基、丙基、丁基、异丙基、叔丁基、苯基、1-萘基、2-萘基、甲氧基、乙氧基、羟基、巯基、氨基、甲氨基、乙氨基、二甲氨基、1-羟基乙基、硝基、氰基、羧基、甲氧基羰基、苄基、氟、氯、溴或碘,m1为钴(ii)、锰(ii)、铁(ii)中的任意一种,m2为铜(ii)、锌(ii)、镍(ii)中的任意一种。
一种非均相双金属中心金属卟啉催化剂的制备方法,按如下方法制备得到:
将卟啉m1(ii)、卟啉m2(ii)、碳酸钾、碘化钾、杂化硅载体置于n,n-二甲基甲酰胺中,搅拌并在氮气气氛、温度为50~200℃(优选80~150℃)的条件下,进行固载化反应1.0~120h(优选24~96h),之后反应体系经抽滤、离心,得到固体物质,将所得固体物质在60~120℃下真空干燥8~36h,得到所述非均相双金属中心金属卟啉催化剂;
所述卟啉m1(ii)与杂化硅载体的质量比为1︰(10~40)(优选1︰(15~30));所述卟啉m2(ii)与卟啉m2(ii)的物质的量之比为1︰(0.5~10)(优选1︰(1~5));所述卟啉m1(ii)与碳酸钾的物质的量之比为1︰(2~20)(优选1︰(5~15));所述卟啉m1(ii)与碘化钾的物质的量之比为1︰(0.5~10)(优选1︰(1~5));所述n,n-二甲基甲酰胺体积用量以杂化硅载体的质量计为5~15ml/g。
进一步,所述卟啉m1(ii)优选为:5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉钴(ii)、5,10,15-三(p-甲氧基)-20-(p-羟基苯基)卟啉钴(ii)、5,10,15-三(p-乙氧基)-20-(p-羟基苯基)卟啉钴(ii)、5,10,15-三(p-溴)-20-(p-羟基苯基)卟啉钴(ii)、5,10,15-三(p-甲基)-20-(p-羟基苯基)卟啉钴(ii)、5,10,15-三(p-羧基)-20-(p-羟基苯基)卟啉钴(ii)或5,10,15-三(p-氟苯基)-20-(p-羟基苯基)卟啉钴(ii)。
再进一步,所述卟啉m2(ii)优选为:5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉锌(ii)、5,10,15-三(p-甲氧基)-20-(p-羟基苯基)卟啉锌(ii)、5,10,15-三(p-乙氧基)-20-(p-羟基苯基)卟啉锌(ii)、5,10,15-三(p-溴)-20-(p-羟基苯基)卟啉锌(ii)、5,10,15-三(p-甲基)-20-(p-羟基苯基)卟啉锌(ii)、5,10,15-三(p-羧基)-20-(p-羟基苯基)卟啉锌(ii)或5,10,15-三(p-氟苯基)-20-(p-羟基苯基)卟啉锌(ii)。
本发明所述杂化硅载体可根据现有文献所报道的方法进行制备,具体可参见journalofmolecularcatallysisa-chemical2010,319,58;europeanjournaloforganicchemistry2012,3625;reactivefunctionalpolymers2013,73,192,即分子催化化学杂志2010,319,58;欧洲有机化学杂志2012,3625;反应性和功能性聚合物2013,73,192。
本发明还提供了一种所述非均相双金属中心金属卟啉催化剂在环烷烃催化氧化反应中的应用。
所述应用的方法为:将非均相双金属卟啉分散于环烷烃中,密封反应体系,搅拌下升温至100~130℃,通入氧气至0.2~3.0mpa,保持设定的温度和氧气压力,搅拌反应3~24h,之后反应液经后处理,得到产物环烷醇和环烷酮。
本发明所述方法中,环烷烃为反应原料,非均相双金属中心金属卟啉为催化剂,分子氧为氧化剂;
所述非均相双金属中心金属卟啉与环烷烃的质量比为1︰100~10000,优选1︰400~850;
所述搅拌的速率为100~1500rpm,优选600~1200rpm;
所述反应温度为100~150℃,优选110~130℃;
所述反应压力0.2~3.0mpa,优选0.6~1.6mpa;
所述反应时间2.0~48h,优选3.0~24h。
所述后处理的方法为:反应结束后,向反应液中加入三苯基膦pph3,用量为环烷烃物质的量的2.5~25%,室温(20~30℃)下搅拌30min还原生成的过氧化物,粗产物经蒸馏,减压精馏和重结晶,得氧化产物。
本发明的技术构思为:本发明以一种非均相双金属中心金属卟啉为催化剂,催化分子氧在无溶剂的条件下氧化环烷烃,不仅显著提高了环烷醇和环烷酮的选择性,环烷烃的转化率也有所提高,实现了环烷烃氧化过程中脂肪族二酸的抑制。因此,本发明以一种非均相双金属卟啉为催化剂催化分子氧在无溶剂的条件下氧化环烷烃,具有解决工业上环烷烃催化氧化过程中,环烷醇和环烷酮易于深度氧化生成脂肪族二酸的潜力,具有重要的工业应用价值和理论研究价值,对其它催化氧化体系选择性的提高也具有一定的参考价值。
本发明的有益效果主要体现在:本发明以非均相双金属中心金属卟啉为催化剂,结构新颖,合成路线简单,应用性广,在环烷烃催化氧化反应中,环烷醇和环烷酮选择性高,有效抑制了脂肪族二酸的生成;脂肪族二酸选择性低,也有利于环烷烃氧化过程的连续化和产品的分离;具有解决工业上环烷烃催化氧化过程中,环烷醇和环烷酮易于深度氧化生成脂肪族二酸的潜力。本发明是一种高效、可行的环烷烃选择性催化氧化新方法。
(四)附图说明
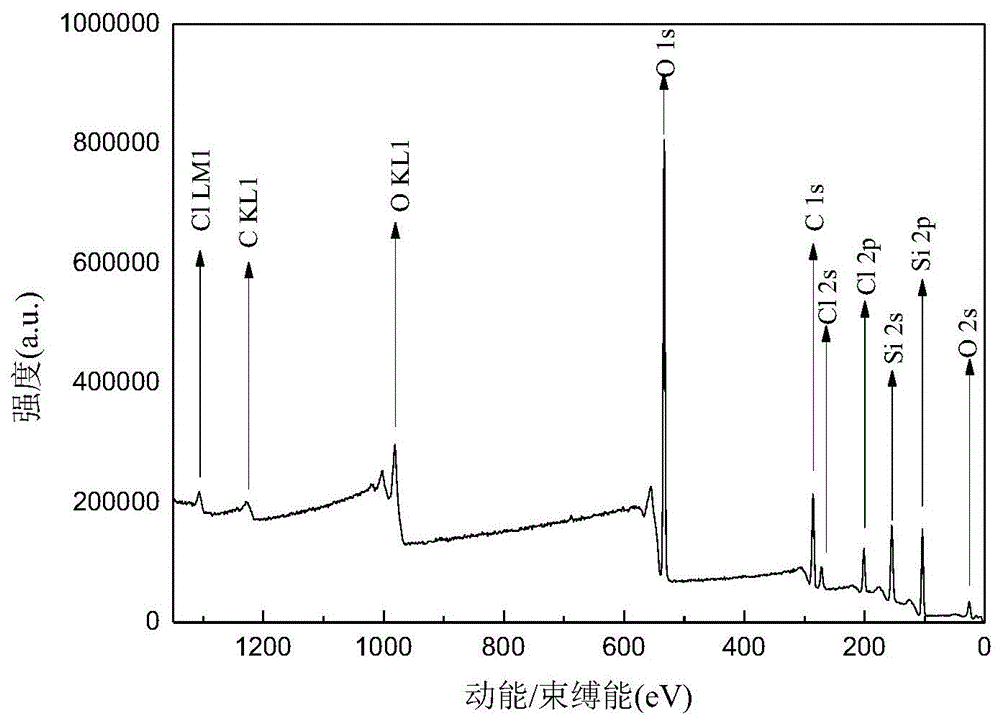
图1是杂化硅载体的xps图谱;
图2是非均相双金属中心金属卟啉的xps图谱;
图3是杂化硅载体(a)、非均相双金属中心金属卟啉(b)的红外图谱。
(五)具体实施方式
下面结合具体实施例对本发明进行进一步说明,但本发明的保护范围并不仅限于此。
实施例1-26是所述非均相双金属中心金属卟啉催化剂的合成;
实施例27-53是所述非均相双金属中心金属卟啉催化剂在环烷烃催化氧化反应中的应用;
实施例54-57是所述非均相双金属中心金属卟啉催化剂在环烷烃催化氧化反应中应用的对比实验;
实施例58是所述非均相双金属中心金属卟啉催化剂在环烷烃催化氧化反应中应用的放大实验。
本发明所述杂化硅载体的制备方法可参见journalofmolecularcatallysisa-chemical2010,319,58;europeanjournaloforganicchemistry2012,3625;reactivefunctionalpolymers2013,73,192,具体为:将12.04g3-氯丙基三乙氧基硅烷、41.67g正硅酸乙酯溶于150ml无水乙醇中,制得溶液①;将17.12g水,20.0ml1m四丁基氟化铵的四氢呋喃溶液溶于100ml无水乙醇中,制得溶液②;将溶液①迅速倒入溶液②,剧烈摇晃10s,静置,室温下老化6.0d。所得固体经无水乙醇洗(3×150ml)后,于80℃下真空干燥10h,得白色固体粉末(杂化硅载体)46.58g。
本发明所用金属卟啉均参考journaloftheamericanchemicalsociety2017,139:18590-18597;journaloftheamericanchemicalsociety2018,140:6383-6390合成。所用试剂均为市售分析纯。
实施例1
在50ml单口反应管中,将2.0000g杂化硅载体、0.1184g(0.15mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉钴(ii)、0.2400g(0.3mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉锌(ii)、0.1866g(1.35mmol)k2co3和0.0500g(0.3mmol)ki置于20mln,n-二甲基甲酰胺中,于氮气气氛室温下搅拌10min后,将反应混合物搅拌升温到50℃,搅拌反应72.0h。冷却到室温,抽滤,转移至10ml离心管中,离心10min,取下层固体,干燥dmf洗涤(10×8ml)至上层液澄清,蒸馏水洗涤(2×8ml)至上层液澄清,干燥丙酮洗涤(5×8ml)至上层液澄清,取下层固体,于80℃干燥12.0h。得棕红色固体粉末(si@p-cl-porp.co&zn)0.3089g。杂化硅载体的xps图谱如附图1所示,非均相双金属中心金属卟啉的xps图谱如附图2所示,杂化硅载体(a)、非均相双金属中心金属卟啉(b)的红外图谱如附图3所示。
实施例2
在50ml单口反应管中,将2.0000g杂化硅载体、0.1184g(0.15mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉钴(ii)、0.2400g(0.3mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉锌(ii)、0.1866g(1.35mmol)k2co3和0.0500g(0.3mmol)ki置于20mln,n-二甲基甲酰胺中,于氮气气氛室温下搅拌10min后,将反应混合物搅拌升温到120℃,搅拌反应72.0h。冷却到室温,抽滤,转移至10ml离心管中,离心10min,取下层固体,干燥dmf洗涤(10×8ml)至上层液澄清,蒸馏水洗涤(2×8ml)至上层液澄清,干燥丙酮洗涤(5×8ml)至上层液澄清,取下层固体,于80℃干燥12.0h。得棕红色固体粉末(si@p-cl-porp.co&zn)0.6039g。
实施例3
在50ml单口反应管中,将2.0000g杂化硅载体、0.1184g(0.15mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉钴(ii)、0.2400g(0.3mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉锌(ii)、0.1866g(1.35mmol)k2co3和0.0500g(0.3mmol)ki置于20mln,n-二甲基甲酰胺中,于氮气气氛室温下搅拌10min后,将反应混合物搅拌升温到200℃,搅拌反应72.0h。冷却到室温,抽滤,转移至10ml离心管中,离心10min,取下层固体,干燥dmf洗涤(10×8ml)至上层液澄清,蒸馏水洗涤(2×8ml)至上层液澄清,干燥丙酮洗涤(5×8ml)至上层液澄清,取下层固体,于80℃干燥12.0h。得棕红色固体粉末(si@p-cl-porp.co&zn)0.7573g。
实施例4
在50ml单口反应管中,将2.0000g杂化硅载体、0.1184g(0.15mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉钴(ii)、0.2400g(0.3mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉锌(ii)、0.1866g(1.35mmol)k2co3和0.0500g(0.3mmol)ki置于20mln,n-二甲基甲酰胺中,于氮气气氛室温下搅拌10min后,将反应混合物搅拌升温到120℃,搅拌反应12.0h。冷却到室温,抽滤,转移至10ml离心管中,离心10min,取下层固体,干燥dmf洗涤(10×8ml)至上层液澄清,蒸馏水洗涤(2×8ml)至上层液澄清,干燥丙酮洗涤(5×8ml)至上层液澄清,取下层固体,于80℃干燥12.0h。得棕红色固体粉末(si@p-cl-porp.co&zn)0.2796g。
实施例5
在50ml单口反应管中,将2.0000g杂化硅载体、0.1184g(0.15mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉钴(ii)、0.2400g(0.3mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉锌(ii)、0.1866g(1.35mmol)k2co3和0.0500g(0.3mmol)ki置于20mln,n-二甲基甲酰胺中,于氮气气氛室温下搅拌10min后,将反应混合物搅拌升温到120℃,搅拌反应96.0h。冷却到室温,抽滤,转移至10ml离心管中,离心10min,取下层固体,干燥dmf洗涤(10×8ml)至上层液澄清,蒸馏水洗涤(2×8ml)至上层液澄清,干燥丙酮洗涤(5×8ml)至上层液澄清,取下层固体,于80℃干燥12.0h。得棕红色固体粉末(si@p-cl-porp.co&zn)0.7845g。
实施例6
在50ml单口反应管中,将2.0000g杂化硅载体、0.1184g(0.15mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉钴(ii)、0.2400g(0.3mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉锌(ii)、0.1866g(1.35mmol)k2co3和0.0500g(0.3mmol)ki置于20mln,n-二甲基甲酰胺中,于氮气气氛室温下搅拌10min后,将反应混合物搅拌升温到120℃,搅拌反应120.0h。冷却到室温,抽滤,转移至10ml离心管中,离心10min,取下层固体,干燥dmf洗涤(10×8ml)至上层液澄清,蒸馏水洗涤(2×8ml)至上层液澄清,干燥丙酮洗涤(5×8ml)至上层液澄清,取下层固体,于80℃干燥12.0h。得棕红色固体粉末(si@p-cl-porp.co&zn)0.8039g。
实施例7
在50ml单口反应管中,将1.1840g杂化硅载体、0.1184g(0.15mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉钴(ii)、0.2400g(0.3mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉锌(ii)、0.1866g(1.35mmol)k2co3和0.0500g(0.3mmol)ki置于20mln,n-二甲基甲酰胺中,于氮气气氛室温下搅拌10min后,将反应混合物搅拌升温到120℃,搅拌反应72.0h。冷却到室温,抽滤,转移至10ml离心管中,离心10min,取下层固体,干燥dmf洗涤(10×8ml)至上层液澄清,蒸馏水洗涤(2×8ml)至上层液澄清,干燥丙酮洗涤(5×8ml)至上层液澄清,取下层固体,于80℃干燥12.0h。得棕红色固体粉末(si@p-cl-porp.co&zn)0.4085g。
实施例8
在50ml单口反应管中,将3.5520g杂化硅载体、0.1184g(0.15mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉钴(ii)、0.2400g(0.3mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉锌(ii)、0.1866g(1.35mmol)k2co3和0.0500g(0.3mmol)ki置于20mln,n-二甲基甲酰胺中,于氮气气氛室温下搅拌10min后,将反应混合物搅拌升温到120℃,搅拌反应72.0h。冷却到室温,抽滤,转移至10ml离心管中,离心10min,取下层固体,干燥dmf洗涤(10×8ml)至上层液澄清,蒸馏水洗涤(2×8ml)至上层液澄清,干燥丙酮洗涤(5×8ml)至上层液澄清,取下层固体,于80℃干燥12.0h。得棕红色固体粉末(si@p-cl-porp.co&zn)0.7123g。
实施例9
在50ml单口反应管中,将4.7360g杂化硅载体、0.1184g(0.15mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉钴(ii)、0.2400g(0.3mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉锌(ii)、0.1866g(1.35mmol)k2co3和0.0500g(0.3mmol)ki置于20mln,n-二甲基甲酰胺中,于氮气气氛室温下搅拌10min后,将反应混合物搅拌升温到120℃,搅拌反应72.0h。冷却到室温,抽滤,转移至10ml离心管中,离心10min,取下层固体,干燥dmf洗涤(10×8ml)至上层液澄清,蒸馏水洗涤(2×8ml)至上层液澄清,干燥丙酮洗涤(5×8ml)至上层液澄清,取下层固体,于80℃干燥12.0h。得棕红色固体粉末(si@p-cl-porp.co&zn)0.8235g。
实施例10
在50ml单口反应管中,将2.0000g杂化硅载体、0.1184g(0.15mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉钴(ii)、0.0592g(0.075mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉锌(ii)、0.1866g(1.35mmol)k2co3和0.0500g(0.3mmol)ki置于20mln,n-二甲基甲酰胺中,于氮气气氛室温下搅拌10min后,将反应混合物搅拌升温到120℃,搅拌反应72.0h。冷却到室温,抽滤,转移至10ml离心管中,离心10min,取下层固体,干燥dmf洗涤(10×8ml)至上层液澄清,蒸馏水洗涤(2×8ml)至上层液澄清,干燥丙酮洗涤(5×8ml)至上层液澄清,取下层固体,于80℃干燥12.0h。得棕红色固体粉末(si@p-cl-porp.co&zn)0.4215g。
实施例11
在50ml单口反应管中,将2.0000g杂化硅载体、0.1184g(0.15mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉钴(ii)、0.5918g(0.75mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉锌(ii)、0.1866g(1.35mmol)k2co3和0.0500g(0.3mmol)ki置于20mln,n-二甲基甲酰胺中,于氮气气氛室温下搅拌10min后,将反应混合物搅拌升温到120℃,搅拌反应72.0h。冷却到室温,抽滤,转移至10ml离心管中,离心10min,取下层固体,干燥dmf洗涤(10×8ml)至上层液澄清,蒸馏水洗涤(2×8ml)至上层液澄清,干燥丙酮洗涤(5×8ml)至上层液澄清,取下层固体,于80℃干燥12.0h。得棕红色固体粉末(si@p-cl-porp.co&zn)0.7764g。
实施例12
在50ml单口反应管中,将2.0000g杂化硅载体、0.1184g(0.15mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉钴(ii)、1.1835g(1.5mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉锌(ii)、0.1866g(1.35mmol)k2co3和0.0500g(0.3mmol)ki置于20mln,n-二甲基甲酰胺中,于氮气气氛室温下搅拌10min后,将反应混合物搅拌升温到120℃,搅拌反应72.0h。冷却到室温,抽滤,转移至10ml离心管中,离心10min,取下层固体,干燥dmf洗涤(10×8ml)至上层液澄清,蒸馏水洗涤(2×8ml)至上层液澄清,干燥丙酮洗涤(5×8ml)至上层液澄清,取下层固体,于80℃干燥12.0h。得棕红色固体粉末(si@p-cl-porp.co&zn)0.9016g。
实施例13
在50ml单口反应管中,将2.0000g杂化硅载体、0.1184g(0.15mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉钴(ii)、0.2400g(0.3mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉锌(ii)、0.0415g(0.3mmol)k2co3和0.0500g(0.3mmol)ki置于20mln,n-二甲基甲酰胺中,于氮气气氛室温下搅拌10min后,将反应混合物搅拌升温到120℃,搅拌反应72.0h。冷却到室温,抽滤,转移至10ml离心管中,离心10min,取下层固体,干燥dmf洗涤(10×8ml)至上层液澄清,蒸馏水洗涤(2×8ml)至上层液澄清,干燥丙酮洗涤(5×8ml)至上层液澄清,取下层固体,于80℃干燥12.0h。得棕红色固体粉末(si@p-cl-porp.co&zn)0.4023g。
实施例14
在50ml单口反应管中,将2.0000g杂化硅载体、0.1184g(0.15mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉钴(ii)、0.2400g(0.3mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉锌(ii)、0.3110g(2.25mmol)k2co3和0.0500g(0.3mmol)ki置于20mln,n-二甲基甲酰胺中,于氮气气氛室温下搅拌10min后,将反应混合物搅拌升温到120℃,搅拌反应72.0h。冷却到室温,抽滤,转移至10ml离心管中,离心10min,取下层固体,干燥dmf洗涤(10×8ml)至上层液澄清,蒸馏水洗涤(2×8ml)至上层液澄清,干燥丙酮洗涤(5×8ml)至上层液澄清,取下层固体,于80℃干燥12.0h。得棕红色固体粉末(si@p-cl-porp.co&zn)0.7011g。
实施例15
在50ml单口反应管中,将2.0000g杂化硅载体、0.1184g(0.15mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉钴(ii)、0.2400g(0.3mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉锌(ii)、0.4146g(3.0mmol)k2co3和0.0500g(0.3mmol)ki置于20mln,n-二甲基甲酰胺中,于氮气气氛室温下搅拌10min后,将反应混合物搅拌升温到120℃,搅拌反应72.0h。冷却到室温,抽滤,转移至10ml离心管中,离心10min,取下层固体,干燥dmf洗涤(10×8ml)至上层液澄清,蒸馏水洗涤(2×8ml)至上层液澄清,干燥丙酮洗涤(5×8ml)至上层液澄清,取下层固体,于80℃干燥12.0h。得棕红色固体粉末(si@p-cl-porp.co&zn)0.7934g。
实施例16
在50ml单口反应管中,将2.0000g杂化硅载体、0.1184g(0.15mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉钴(ii)、0.2400g(0.3mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉锌(ii)、0.1866g(1.35mmol)k2co3和0.0125g(0.075mmol)ki置于20mln,n-二甲基甲酰胺中,于氮气气氛室温下搅拌10min后,将反应混合物搅拌升温到120℃,搅拌反应72.0h。冷却到室温,抽滤,转移至10ml离心管中,离心10min,取下层固体,干燥dmf洗涤(10×8ml)至上层液澄清,蒸馏水洗涤(2×8ml)至上层液澄清,干燥丙酮洗涤(5×8ml)至上层液澄清,取下层固体,于80℃干燥12.0h。得棕红色固体粉末(si@p-cl-porp.co&zn)0.5467g。
实施例17
在50ml单口反应管中,将2.0000g杂化硅载体、0.1184g(0.15mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉钴(ii)、0.2400g(0.3mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉锌(ii)、0.1866g(1.35mmol)k2co3和0.1245g(0.75mmol)ki置于20mln,n-二甲基甲酰胺中,于氮气气氛室温下搅拌10min后,将反应混合物搅拌升温到120℃,搅拌反应72.0h。冷却到室温,抽滤,转移至10ml离心管中,离心10min,取下层固体,干燥dmf洗涤(10×8ml)至上层液澄清,蒸馏水洗涤(2×8ml)至上层液澄清,干燥丙酮洗涤(5×8ml)至上层液澄清,取下层固体,于80℃干燥12.0h。得棕红色固体粉末(si@p-cl-porp.co&zn)0.6839g。
实施例18
在50ml单口反应管中,将2.0000g杂化硅载体、0.1184g(0.15mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉钴(ii)、0.2400g(0.3mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉锌(ii)、0.1866g(1.35mmol)k2co3和0.2490g(1.5mmol)ki置于20mln,n-二甲基甲酰胺中,于氮气气氛室温下搅拌10min后,将反应混合物搅拌升温到120℃,搅拌反应72.0h。冷却到室温,抽滤,转移至10ml离心管中,离心10min,取下层固体,干燥dmf洗涤(10×8ml)至上层液澄清,蒸馏水洗涤(2×8ml)至上层液澄清,干燥丙酮洗涤(5×8ml)至上层液澄清,取下层固体,于80℃干燥12.0h。得棕红色固体粉末(si@p-cl-porp.co&zn)0.7452g。
实施例19
在50ml单口反应管中,将2.0000g杂化硅载体、0.1184g(0.15mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉钴(ii)、0.2400g(0.3mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉锌(ii)、0.1866g(1.35mmol)k2co3和0.0500g(0.3mmol)ki置于10mln,n-二甲基甲酰胺中,于氮气气氛室温下搅拌10min后,将反应混合物搅拌升温到120℃,搅拌反应72.0h。冷却到室温,抽滤,转移至10ml离心管中,离心10min,取下层固体,干燥dmf洗涤(10×8ml)至上层液澄清,蒸馏水洗涤(2×8ml)至上层液澄清,干燥丙酮洗涤(5×8ml)至上层液澄清,取下层固体,于80℃干燥12.0h。得棕红色固体粉末(si@p-cl-porp.co&zn)0.4958g。
实施例20
在50ml单口反应管中,将2.0000g杂化硅载体、0.1184g(0.15mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉钴(ii)、0.2400g(0.3mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉锌(ii)、0.1866g(1.35mmol)k2co3和0.0500g(0.3mmol)ki置于30mln,n-二甲基甲酰胺中,于氮气气氛室温下搅拌10min后,将反应混合物搅拌升温到120℃,搅拌反应72.0h。冷却到室温,抽滤,转移至10ml离心管中,离心10min,取下层固体,干燥dmf洗涤(10×8ml)至上层液澄清,蒸馏水洗涤(2×8ml)至上层液澄清,干燥丙酮洗涤(5×8ml)至上层液澄清,取下层固体,于80℃干燥12.0h。得棕红色固体粉末(si@p-cl-porp.co&zn)0.6113g。
实施例21
在50ml单口反应管中,将2.0000g杂化硅载体、0.1386g(0.15mmol)5,10,15-三(p-溴苯基)-20-(p-羟基苯基)卟啉钴(ii)、0.2772g(0.3mmol)5,10,15-三(p-溴苯基)-20-(p-羟基苯基)卟啉锌(ii)、0.1866g(1.35mmol)k2co3和0.0500g(0.3mmol)ki置于20mln,n-二甲基甲酰胺中,于氮气气氛室温下搅拌10min后,将反应混合物搅拌升温到120℃,搅拌反应72.0h。冷却到室温,抽滤,转移至10ml离心管中,离心10min,取下层固体,干燥dmf洗涤(10×8ml)至上层液澄清,蒸馏水洗涤(2×8ml)至上层液澄清,干燥丙酮洗涤(5×8ml)至上层液澄清,取下层固体,于80℃干燥12.0h。得棕红色固体粉末(si@p-br-porp.co&zn)0.7041g。
实施例22
在50ml单口反应管中,将2.0000g杂化硅载体、0.1098g(0.15mmol)5,10,15-三(p-氨基)-20-(p-羟基苯基)卟啉钴(ii)、0.2196g(0.3mmol)5,10,15-三(p-氨基)-20-(p-羟基苯基)卟啉锌(ii)、0.1866g(1.35mmol)k2co3和0.0500g(0.3mmol)ki置于20mln,n-二甲基甲酰胺中,于氮气气氛室温下搅拌10min后,将反应混合物搅拌升温到120℃,搅拌反应72.0h。冷却到室温,抽滤,转移至10ml离心管中,离心10min,取下层固体,干燥dmf洗涤(10×8ml)至上层液澄清,蒸馏水洗涤(2×8ml)至上层液澄清,干燥丙酮洗涤(5×8ml)至上层液澄清,取下层固体,于80℃干燥12.0h。得棕红色固体粉末(si@p-nh2-porp.co&zn)0.5883g。
实施例23
在50ml单口反应管中,将2.0000g杂化硅载体、0.1098g(0.15mmol)5,10,15-三(p-甲基)-20-(p-羟基苯基)卟啉钴(ii)、0.2196g(0.3mmol)5,10,15-三(p-甲基)-20-(p-羟基苯基)卟啉锌(ii)、0.1866g(1.35mmol)k2co3和0.0500g(0.3mmol)ki置于20mln,n-二甲基甲酰胺中,于氮气气氛室温下搅拌10min后,将反应混合物搅拌升温到120℃,搅拌反应72.0h。冷却到室温,抽滤,转移至10ml离心管中,离心10min,取下层固体,干燥dmf洗涤(10×8ml)至上层液澄清,蒸馏水洗涤(2×8ml)至上层液澄清,干燥丙酮洗涤(5×8ml)至上层液澄清,取下层固体,于80℃干燥12.0h。得棕红色固体粉末(si@p-ch3-porp.co&zn)0.5746g。
实施例24
在50ml单口反应管中,将2.0000g杂化硅载体、0.1184g(0.15mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉钴(ii)、0.2385g(0.3mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉铜(ii)、0.1866g(1.35mmol)k2co3和0.0500g(0.3mmol)ki置于20mln,n-二甲基甲酰胺中,于氮气气氛室温下搅拌10min后,将反应混合物搅拌升温到120℃,搅拌反应72.0h。冷却到室温,抽滤,转移至10ml离心管中,离心10min,取下层固体,干燥dmf洗涤(10×8ml)至上层液澄清,蒸馏水洗涤(2×8ml)至上层液澄清,干燥丙酮洗涤(5×8ml)至上层液澄清,取下层固体,于80℃干燥12.0h。得棕红色固体粉末(si@p-cl-porp.co&cu)0.6178g。
实施例25
在50ml单口反应管中,将2.0000g杂化硅载体、0.1179g(0.15mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉锰(ii)、0.2400g(0.3mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉锌(ii)、0.1866g(1.35mmol)k2co3和0.0500g(0.3mmol)ki置于20mln,n-二甲基甲酰胺中,于氮气气氛室温下搅拌10min后,将反应混合物搅拌升温到120℃,搅拌反应72.0h。冷却到室温,抽滤,转移至10ml离心管中,离心10min,取下层固体,干燥dmf洗涤(10×8ml)至上层液澄清,蒸馏水洗涤(2×8ml)至上层液澄清,干燥丙酮洗涤(5×8ml)至上层液澄清,取下层固体,于80℃干燥12.0h。得棕红色固体粉末(si@p-cl-porp.mn&zn)0.5939g。
实施例26
在50ml单口反应管中,将2.0000g杂化硅载体、0.1184g(0.15mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉铁(ii)、0.2370g(0.3mmol)5,10,15-三(p-氯苯基)-20-(p-羟基苯基)卟啉镍(ii)、0.1866g(1.35mmol)k2co3和0.0500g(0.3mmol)ki置于20mln,n-二甲基甲酰胺中,于氮气气氛室温下搅拌10min后,将反应混合物搅拌升温到120℃,搅拌反应72.0h。冷却到室温,抽滤,转移至10ml离心管中,离心10min,取下层固体,干燥dmf洗涤(10×8ml)至上层液澄清,蒸馏水洗涤(2×8ml)至上层液澄清,干燥丙酮洗涤(5×8ml)至上层液澄清,取下层固体,于80℃干燥12.0h。得棕红色固体粉末(si@p-cl-porp.fe&ni)0.6112g。
实施例27
在100ml具有聚四氟乙烯内胆的不锈钢高压反应釜中,将0.0200gsi@p-cl-porp.co&zn分散于16.8320g(200mmol)环己烷中,密封反应釜,搅拌升温到120℃,通入氧气至1.0mpa。于120℃,1.0mpa氧气压力,800rpm搅拌反应8.0h。反应完毕,冰水冷却至室温,向反应混合物中加入1.3115g(5.00mmol)三苯基膦(pph3),室温下搅拌30min还原生成的过氧化物。以丙酮为溶剂,将所得反应混合物定容至100ml。移取10ml所得溶液,以甲苯为内标,进行气相色谱分析;移取10ml所得溶液,以苯甲酸为内标,进行液相色谱分析。环己烷转化率3.57%,环己醇选择性48.9%,环己酮选择性42.8%,环己基过氧化氢选择性5.3%,己二酸选择性2.1%,戊二酸选择性0.9%。
实施例28
在100ml具有聚四氟乙烯内胆的不锈钢高压反应釜中,将0.0400gsi@p-cl-porp.co&zn分散于16.8320g(200mmol)环己烷中,密封反应釜,搅拌升温到120℃,通入氧气至1.0mpa。于120℃,1.0mpa氧气压力,800rpm搅拌反应8.0h。反应完毕,冰水冷却至室温,向反应混合物中加入1.3115g(5.00mmol)三苯基膦(pph3),室温下搅拌30min还原生成的过氧化物。以丙酮为溶剂,将所得反应混合物定容至100ml。移取10ml所得溶液,以甲苯为内标,进行气相色谱分析;移取10ml所得溶液,以苯甲酸为内标,进行液相色谱分析。环己烷转化率5.57%,环己醇选择性51.6%,环己酮选择性43.7%,环己基过氧化氢选择性2.9%,己二酸选择性1.1%,戊二酸选择性0.7%。
实施例29
在100ml具有聚四氟乙烯内胆的不锈钢高压反应釜中,将0.0600gsi@p-cl-porp.co&zn分散于16.8320g(200mmol)环己烷中,密封反应釜,搅拌升温到120℃,通入氧气至1.0mpa。于120℃,1.0mpa氧气压力,800rpm搅拌反应8.0h。反应完毕,冰水冷却至室温,向反应混合物中加入1.3115g(5.00mmol)三苯基膦(pph3),室温下搅拌30min还原生成的过氧化物。以丙酮为溶剂,将所得反应混合物定容至100ml。移取10ml所得溶液,以甲苯为内标,进行气相色谱分析;移取10ml所得溶液,以苯甲酸为内标,进行液相色谱分析。环己烷转化率5.08%,环己醇选择性48.2%,环己酮选择性46.0%,环己基过氧化氢选择性3.4%,己二酸选择性1.5%,戊二酸选择性0.9%。
实施例30
在100ml具有聚四氟乙烯内胆的不锈钢高压反应釜中,将0.0400gsi@p-cl-porp.co&zn分散于16.8320g(200mmol)环己烷中,密封反应釜,搅拌升温到120℃,通入氧气至0.2mpa。于120℃,0.2mpa氧气压力,800rpm搅拌反应8.0h。反应完毕,冰水冷却至室温,向反应混合物中加入1.3115g(5.00mmol)三苯基膦(pph3),室温下搅拌30min还原生成的过氧化物。以丙酮为溶剂,将所得反应混合物定容至100ml。移取10ml所得溶液,以甲苯为内标,进行气相色谱分析;移取10ml所得溶液,以苯甲酸为内标,进行液相色谱分析。环己烷转化率1.57%,环己醇选择性36.1%,环己酮选择性38.9%,环己基过氧化氢选择性16%,己二酸选择性7.6%,戊二酸选择性1.3%。
实施例31
在100ml具有聚四氟乙烯内胆的不锈钢高压反应釜中,将0.0400gsi@p-cl-porp.co&zn分散于16.8320g(200mmol)环己烷中,密封反应釜,搅拌升温到120℃,通入氧气至0.6mpa。于120℃,0.6mpa氧气压力,800rpm搅拌反应8.0h。反应完毕,冰水冷却至室温,向反应混合物中加入1.3115g(5.00mmol)三苯基膦(pph3),室温下搅拌30min还原生成的过氧化物。以丙酮为溶剂,将所得反应混合物定容至100ml。移取10ml所得溶液,以甲苯为内标,进行气相色谱分析;移取10ml所得溶液,以苯甲酸为内标,进行液相色谱分析。环己烷转化率2.97%,环己醇选择性40.7%,环己酮选择性34.0%,环己基过氧化氢选择性20.9%,己二酸选择性3.7%,戊二酸选择性0.6%。
实施例32
在100ml具有聚四氟乙烯内胆的不锈钢高压反应釜中,将0.0400gsi@p-cl-porp.co&zn分散于16.8320g(200mmol)环己烷中,密封反应釜,搅拌升温到120℃,通入氧气至1.2mpa。于120℃,1.2mpa氧气压力,800rpm搅拌反应8.0h。反应完毕,冰水冷却至室温,向反应混合物中加入1.3115g(5.00mmol)三苯基膦(pph3),室温下搅拌30min还原生成的过氧化物。以丙酮为溶剂,将所得反应混合物定容至100ml。移取10ml所得溶液,以甲苯为内标,进行气相色谱分析;移取10ml所得溶液,以苯甲酸为内标,进行液相色谱分析。环己烷转化率5.72%,环己醇选择性51.7%,环己酮选择性44.0%,环己基过氧化氢选择性2.3%,己二酸选择性1.2%,戊二酸选择性0.8%。
实施例33
在100ml具有聚四氟乙烯内胆的不锈钢高压反应釜中,将0.0400gsi@p-cl-porp.co&zn分散于16.8320g(200mmol)环己烷中,密封反应釜,搅拌升温到120℃,通入氧气至3.0mpa。于120℃,3.0mpa氧气压力,800rpm搅拌反应8.0h。反应完毕,冰水冷却至室温,向反应混合物中加入1.3115g(5.00mmol)三苯基膦(pph3),室温下搅拌30min还原生成的过氧化物。以丙酮为溶剂,将所得反应混合物定容至100ml。移取10ml所得溶液,以甲苯为内标,进行气相色谱分析;移取10ml所得溶液,以苯甲酸为内标,进行液相色谱分析。环己烷转化率3.96%,环己醇选择性43.0%,环己酮选择性42.6%,环己基过氧化氢选择性9.0%,己二酸选择性4.1%,戊二酸选择性1.3%。
实施例34
在100ml具有聚四氟乙烯内胆的不锈钢高压反应釜中,将0.0400gsi@p-cl-porp.co&zn分散于16.8320g(200mmol)环己烷中,密封反应釜,搅拌升温到100℃,通入氧气至1.0mpa。于100℃,1.0mpa氧气压力,800rpm搅拌反应8.0h。反应完毕,冰水冷却至室温,向反应混合物中加入1.3115g(5.00mmol)三苯基膦(pph3),室温下搅拌30min还原生成的过氧化物。以丙酮为溶剂,将所得反应混合物定容至100ml。移取10ml所得溶液,以甲苯为内标,进行气相色谱分析;移取10ml所得溶液,以苯甲酸为内标,进行液相色谱分析。环己烷转化率3.63%,环己醇选择性40.9%,环己酮选择性39.1%,环己基过氧化氢选择性11.0%,己二酸选择性6.5%,戊二酸选择性2.5%。
实施例35
在100ml具有聚四氟乙烯内胆的不锈钢高压反应釜中,将0.0400gsi@p-cl-porp.co&zn分散于16.8320g(200mmol)环己烷中,密封反应釜,搅拌升温到110℃,通入氧气至1.0mpa。于110℃,1.0mpa氧气压力,800rpm搅拌反应8.0h。反应完毕,冰水冷却至室温,向反应混合物中加入1.3115g(5.00mmol)三苯基膦(pph3),室温下搅拌30min还原生成的过氧化物。以丙酮为溶剂,将所得反应混合物定容至100ml。移取10ml所得溶液,以甲苯为内标,进行气相色谱分析;移取10ml所得溶液,以苯甲酸为内标,进行液相色谱分析。环己烷转化率3.78%,环己醇选择性45.7%,环己酮选择性36.7%,环己基过氧化氢选择性9.6%,己二酸选择性5.8%,戊二酸选择性2.2%。
实施例36
在100ml具有聚四氟乙烯内胆的不锈钢高压反应釜中,将0.0400gsi@p-cl-porp.co&zn分散于16.8320g(200mmol)环己烷中,密封反应釜,搅拌升温到130℃,通入氧气至1.0mpa。于130℃,1.0mpa氧气压力,800rpm搅拌反应8.0h。反应完毕,冰水冷却至室温,向反应混合物中加入1.3115g(5.00mmol)三苯基膦(pph3),室温下搅拌30min还原生成的过氧化物。以丙酮为溶剂,将所得反应混合物定容至100ml。移取10ml所得溶液,以甲苯为内标,进行气相色谱分析;移取10ml所得溶液,以苯甲酸为内标,进行液相色谱分析。环己烷转化率6.13%,环己醇选择性51.6%,环己酮选择性44.3%,环己基过氧化氢选择性2.3%,己二酸选择性1.1%,戊二酸选择性0.7%。
实施例37
在100ml具有聚四氟乙烯内胆的不锈钢高压反应釜中,将0.0400gsi@p-cl-porp.co&zn分散于16.8320g(200mmol)环己烷中,密封反应釜,搅拌升温到120℃,通入氧气至1.0mpa。于120℃,1.0mpa氧气压力,800rpm搅拌反应3.0h。反应完毕,冰水冷却至室温,向反应混合物中加入1.3115g(5.00mmol)三苯基膦(pph3),室温下搅拌30min还原生成的过氧化物。以丙酮为溶剂,将所得反应混合物定容至100ml。移取10ml所得溶液,以甲苯为内标,进行气相色谱分析;移取10ml所得溶液,以苯甲酸为内标,进行液相色谱分析。环己烷转化率1.28%,环己醇选择性30.4%,环己酮选择性28.6%,环己基过氧化氢选择性29.1%,己二酸选择性7.6%,戊二酸选择性4.3%。
实施例38
在100ml具有聚四氟乙烯内胆的不锈钢高压反应釜中,将0.0400gsi@p-cl-porp.co&zn分散于16.8320g(200mmol)环己烷中,密封反应釜,搅拌升温到120℃,通入氧气至1.0mpa。于120℃,1.0mpa氧气压力,800rpm搅拌反应12.0h。反应完毕,冰水冷却至室温,向反应混合物中加入1.3115g(5.00mmol)三苯基膦(pph3),室温下搅拌30min还原生成的过氧化物。以丙酮为溶剂,将所得反应混合物定容至100ml。移取10ml所得溶液,以甲苯为内标,进行气相色谱分析;移取10ml所得溶液,以苯甲酸为内标,进行液相色谱分析。环己烷转化率5.86%,环己醇选择性51.6%,环己酮选择性44.1%,环己基过氧化氢选择性2.5%,己二酸选择性1.1%,戊二酸选择性0.7%。
实施例39
在100ml具有聚四氟乙烯内胆的不锈钢高压反应釜中,将0.0400gsi@p-cl-porp.co&zn分散于16.8320g(200mmol)环己烷中,密封反应釜,搅拌升温到120℃,通入氧气至1.0mpa。于120℃,1.0mpa氧气压力,800rpm搅拌反应24.0h。反应完毕,冰水冷却至室温,向反应混合物中加入1.3115g(5.00mmol)三苯基膦(pph3),室温下搅拌30min还原生成的过氧化物。以丙酮为溶剂,将所得反应混合物定容至100ml。移取10ml所得溶液,以甲苯为内标,进行气相色谱分析;移取10ml所得溶液,以苯甲酸为内标,进行液相色谱分析。环己烷转化率5.93%,环己醇选择性51.6%,环己酮选择性44.3%,环己基过氧化氢选择性2.3%,己二酸选择性1.1%,戊二酸选择性0.7%。
实施例40
在100ml具有聚四氟乙烯内胆的不锈钢高压反应釜中,将0.0400gsi@p-cl-porp.co&zn分散于16.8320g(200mmol)环己烷中,密封反应釜,搅拌升温到120℃,通入氧气至1.0mpa。于120℃,1.0mpa氧气压力,600rpm搅拌反应8.0h。反应完毕,冰水冷却至室温,向反应混合物中加入1.3115g(5.00mmol)三苯基膦(pph3),室温下搅拌30min还原生成的过氧化物。以丙酮为溶剂,将所得反应混合物定容至100ml。移取10ml所得溶液,以甲苯为内标,进行气相色谱分析;移取10ml所得溶液,以苯甲酸为内标,进行液相色谱分析。环己烷转化率4.89%,环己醇选择性50.3%,环己酮选择性41.5%,环己基过氧化氢选择性4.8%,己二酸选择性2.3%,戊二酸选择性1.1%。
实施例41
在100ml具有聚四氟乙烯内胆的不锈钢高压反应釜中,将0.0400gsi@p-cl-porp.co&zn分散于16.8320g(200mmol)环己烷中,密封反应釜,搅拌升温到120℃,通入氧气至1.0mpa。于120℃,1.0mpa氧气压力,1200rpm搅拌反应8.0h。反应完毕,冰水冷却至室温,向反应混合物中加入1.3115g(5.00mmol)三苯基膦(pph3),室温下搅拌30min还原生成的过氧化物。以丙酮为溶剂,将所得反应混合物定容至100ml。移取10ml所得溶液,以甲苯为内标,进行气相色谱分析;移取10ml所得溶液,以苯甲酸为内标,进行液相色谱分析。环己烷转化率5.68%,环己醇选择性51.1%,环己酮选择性43.9%,环己基过氧化氢选择性2.7%,己二酸选择性1.3%,戊二酸选择性1.0%。
实施例42
在100ml具有聚四氟乙烯内胆的不锈钢高压反应釜中,将0.0400gsi@p-cl-porp.co&zn分散于16.8320g(200mmol)环己烷中,密封反应釜,搅拌升温到120℃,通入氧气至1.0mpa。于120℃,1.0mpa氧气压力,100rpm搅拌反应8.0h。反应完毕,冰水冷却至室温,向反应混合物中加入1.3115g(5.00mmol)三苯基膦(pph3),室温下搅拌30min还原生成的过氧化物。以丙酮为溶剂,将所得反应混合物定容至100ml。移取10ml所得溶液,以甲苯为内标,进行气相色谱分析;移取10ml所得溶液,以苯甲酸为内标,进行液相色谱分析。环己烷转化率2.89%,环己醇选择性42.6%,环己酮选择性34.7%,环己基过氧化氢选择性14.0%,己二酸选择性7.3%,戊二酸选择性1.4%。
实施例43
在100ml具有聚四氟乙烯内胆的不锈钢高压反应釜中,将0.0400gsi@p-cl-porp.co&zn分散于14.0240g(200mmol)环戊烷中,密封反应釜,搅拌升温到120℃,通入氧气至1.0mpa。于120℃,1.0mpa氧气压力,800rpm搅拌反应8.0h。反应完毕,冰水冷却至室温,向反应混合物中加入1.3115g(5.00mmol)三苯基膦(pph3),室温下搅拌30min还原生成的过氧化物。以丙酮为溶剂,将所得反应混合物定容至100ml。移取10ml所得溶液,以甲苯为内标,进行气相色谱分析;移取10ml所得溶液,以苯甲酸为内标,进行液相色谱分析。环戊烷转化率4.23%,环戊醇选择性43.9%,环戊酮选择性46.0%,环戊基过氧化氢选择性6.2%,戊二酸选择性2.3%,丁二酸选择性1.6%。
实施例44
在100ml具有聚四氟乙烯内胆的不锈钢高压反应釜中,将0.0400gsi@p-cl-porp.co&zn分散于19.6380g(200mmol)环庚烷中,密封反应釜,搅拌升温到120℃,通入氧气至1.0mpa。于120℃,1.0mpa氧气压力,800rpm搅拌反应8.0h。反应完毕,冰水冷却至室温,向反应混合物中加入1.3115g(5.00mmol)三苯基膦(pph3),室温下搅拌30min还原生成的过氧化物。以丙酮为溶剂,将所得反应混合物定容至100ml。移取10ml所得溶液,以甲苯为内标,进行气相色谱分析;移取10ml所得溶液,以苯甲酸为内标,进行液相色谱分析。环庚烷转化率29.41%,环庚醇选择性47.9%,环庚酮选择性45.6%,环庚基过氧化氢选择性4.7%,庚二酸选择性1.8%,未检测到己二酸生成。
实施例45
在100ml具有聚四氟乙烯内胆的不锈钢高压反应釜中,将0.0400gsi@p-cl-porp.co&zn分散于22.4410g(200mmol)环辛烷中,密封反应釜,搅拌升温到120℃,通入氧气至1.0mpa。于120℃,1.0mpa氧气压力,800rpm搅拌反应8.0h。反应完毕,冰水冷却至室温,向反应混合物中加入1.3115g(5.00mmol)三苯基膦(pph3),室温下搅拌30min还原生成的过氧化物。以丙酮为溶剂,将所得反应混合物定容至100ml。移取10ml所得溶液,以甲苯为内标,进行气相色谱分析;移取10ml所得溶液,以苯甲酸为内标,进行液相色谱分析。环辛烷转化率40.67%,环辛醇选择性48.5%,环辛酮选择性46.2%,环辛基过氧化氢选择性3.2%,辛二酸选择性2.1%,未检测到庚二酸生成。
实施例46
在100ml具有聚四氟乙烯内胆的不锈钢高压反应釜中,将0.0400gsi@p-cl-porp.co&zn分散于33.6640g(200mmol)环十二烷中,密封反应釜,搅拌升温到120℃,通入氧气至1.0mpa。于120℃,1.0mpa氧气压力,800rpm搅拌反应8.0h。反应完毕,冰水冷却至室温,向反应混合物中加入1.3115g(5.00mmol)三苯基膦(pph3),室温下搅拌30min还原生成的过氧化物。以丙酮为溶剂,将所得反应混合物定容至100ml。移取10ml所得溶液,以甲苯为内标,进行气相色谱分析;移取10ml所得溶液,以苯甲酸为内标,进行液相色谱分析。环十二烷转化率47.32%,环十二醇选择性50.7%,环十二酮选择性45.3%,环十二基过氧化氢选择性3.3%,十二碳二酸选择性0.7%,未检测到十一碳二酸生成。
实施例47
在100ml具有聚四氟乙烯内胆的不锈钢高压反应釜中,将0.0400gsi@p-och3-porp.co&zn分散于16.8320g(200mmol)环己烷中,密封反应釜,搅拌升温到120℃,通入氧气至1.0mpa。于120℃,1.0mpa氧气压力,800rpm搅拌反应8.0h。反应完毕,冰水冷却至室温,向反应混合物中加入1.3115g(5.00mmol)三苯基膦(pph3),室温下搅拌30min还原生成的过氧化物。以丙酮为溶剂,将所得反应混合物定容至100ml。移取10ml所得溶液,以甲苯为内标,进行气相色谱分析;移取10ml所得溶液,以苯甲酸为内标,进行液相色谱分析。环己烷转化率4.67%,环己醇选择性50.6%,环己酮选择性43.7%,环己基过氧化氢选择性3.5%,己二酸选择性1.3%,戊二酸选择性0.9%。
实施例48
在100ml具有聚四氟乙烯内胆的不锈钢高压反应釜中,将0.0400gsi@p-ch3-porp.co&zn分散于16.8320g(200mmol)环己烷中,密封反应釜,搅拌升温到120℃,通入氧气至1.0mpa。于120℃,1.0mpa氧气压力,800rpm搅拌反应8.0h。反应完毕,冰水冷却至室温,向反应混合物中加入1.3115g(5.00mmol)三苯基膦(pph3),室温下搅拌30min还原生成的过氧化物。以丙酮为溶剂,将所得反应混合物定容至100ml。移取10ml所得溶液,以甲苯为内标,进行气相色谱分析;移取10ml所得溶液,以苯甲酸为内标,进行液相色谱分析。环己烷转化率4.89%,环己醇选择性49.4%,环己酮选择性43.8%,环己基过氧化氢选择性4.0%,己二酸选择性1.9%,戊二酸选择性0.9%。
实施例49
在100ml具有聚四氟乙烯内胆的不锈钢高压反应釜中,将0.0400gsi@p-br-porp.co&zn分散于16.8320g(200mmol)环己烷中,密封反应釜,搅拌升温到120℃,通入氧气至1.0mpa。于120℃,1.0mpa氧气压力,800rpm搅拌反应8.0h。反应完毕,冰水冷却至室温,向反应混合物中加入1.3115g(5.00mmol)三苯基膦(pph3),室温下搅拌30min还原生成的过氧化物。以丙酮为溶剂,将所得反应混合物定容至100ml。移取10ml所得溶液,以甲苯为内标,进行气相色谱分析;移取10ml所得溶液,以苯甲酸为内标,进行液相色谱分析。环己烷转化率5.13%,环己醇选择性49.1%,环己酮选择性45.3%,环己基过氧化氢选择性3.0%,己二酸选择性1.7%,戊二酸选择性0.9%。
实施例50
在100ml具有聚四氟乙烯内胆的不锈钢高压反应釜中,将0.0400gsi@p-nh2-porp.co&zn分散于16.8320g(200mmol)环己烷中,密封反应釜,搅拌升温到120℃,通入氧气至1.0mpa。于120℃,1.0mpa氧气压力,800rpm搅拌反应8.0h。反应完毕,冰水冷却至室温,向反应混合物中加入1.3115g(5.00mmol)三苯基膦(pph3),室温下搅拌30min还原生成的过氧化物。以丙酮为溶剂,将所得反应混合物定容至100ml。移取10ml所得溶液,以甲苯为内标,进行气相色谱分析;移取10ml所得溶液,以苯甲酸为内标,进行液相色谱分析。环己烷转化率4.73%,环己醇选择性48.1%,环己酮选择性45.7%,环己基过氧化氢选择性3.7%,己二酸选择性1.8%,戊二酸选择性0.7%。
实施例51
在100ml具有聚四氟乙烯内胆的不锈钢高压反应釜中,将0.0400gsi@p-cl-porp.co&cu分散于16.8320g(200mmol)环己烷中,密封反应釜,搅拌升温到120℃,通入氧气至1.0mpa。于120℃,1.0mpa氧气压力,800rpm搅拌反应8.0h。反应完毕,冰水冷却至室温,向反应混合物中加入1.3115g(5.00mmol)三苯基膦(pph3),室温下搅拌30min还原生成的过氧化物。以丙酮为溶剂,将所得反应混合物定容至100ml。移取10ml所得溶液,以甲苯为内标,进行气相色谱分析;移取10ml所得溶液,以苯甲酸为内标,进行液相色谱分析。环己烷转化率4.03%,环己醇选择性44.8%,环己酮选择性40.3%,环己基过氧化氢选择性6.7%,己二酸选择性5.6%,戊二酸选择性2.6%。
实施例52
在100ml具有聚四氟乙烯内胆的不锈钢高压反应釜中,将0.0400gsi@p-cl-porp.mn&zn分散于16.8320g(200mmol)环己烷中,密封反应釜,搅拌升温到120℃,通入氧气至1.0mpa。于120℃,1.0mpa氧气压力,800rpm搅拌反应8.0h。反应完毕,冰水冷却至室温,向反应混合物中加入1.3115g(5.00mmol)三苯基膦(pph3),室温下搅拌30min还原生成的过氧化物。以丙酮为溶剂,将所得反应混合物定容至100ml。移取10ml所得溶液,以甲苯为内标,进行气相色谱分析;移取10ml所得溶液,以苯甲酸为内标,进行液相色谱分析。环己烷转化率4.33%,环己醇选择性46.9%,环己酮选择性40.5%,环己基过氧化氢选择性5.4%,己二酸选择性4.6%,戊二酸选择性2.6%。
实施例53
在100ml具有聚四氟乙烯内胆的不锈钢高压反应釜中,将0.0400gsi@p-cl-porp.fe&ni分散于16.8320g(200mmol)环己烷中,密封反应釜,搅拌升温到120℃,通入氧气至1.0mpa。于120℃,1.0mpa氧气压力,800rpm搅拌反应8.0h。反应完毕,冰水冷却至室温,向反应混合物中加入1.3115g(5.00mmol)三苯基膦(pph3),室温下搅拌30min还原生成的过氧化物。以丙酮为溶剂,将所得反应混合物定容至100ml。移取10ml所得溶液,以甲苯为内标,进行气相色谱分析;移取10ml所得溶液,以苯甲酸为内标,进行液相色谱分析。环己烷转化率3.43%,环己醇选择性40.0%,环己酮选择性39.6%,环己基过氧化氢选择性11.0%,己二酸选择性8.1%,戊二酸选择性1.3%。
实施例54(对比实验)
在100ml具有聚四氟乙烯内胆的不锈钢高压反应釜中,将0.0020g(0.0030mmol)5,10,15-三(p-氯苯基)-20-(p-甲氧基苯基)卟啉锌(ii)、0.0020g(0.0030mmol)5,10,15-三(p-氯苯基)-20-(p-甲氧基苯基)卟啉钴(ii)分散于16.8320g(200mmol)环己烷中,密封反应釜,搅拌升温到120℃,通入氧气至1.0mpa。于120℃,1.0mpa氧气压力,800rpm搅拌反应8.0h。反应完毕,冰水冷却至室温,向反应混合物中加入1.3115g(5.00mmol)三苯基膦(pph3),室温下搅拌30min还原生成的过氧化物。以丙酮为溶剂,将所得反应混合物定容至100ml。移取10ml所得溶液,以甲苯为内标,进行气相色谱分析;移取10ml所得溶液,以苯甲酸为内标,进行液相色谱分析。环己烷转化率3.76%,环己醇选择性34.8%,环己酮选择性35.3%,环己基过氧化氢选择性14.1%,己二酸选择性10.4%,戊二酸选择性5.3%。
实施例55(对比实验)
在100ml具有聚四氟乙烯内胆的不锈钢高压反应釜中,将0.0400gsi@p-cl-porp.zn分散于16.8320g(200mmol)环己烷中,密封反应釜,搅拌升温到120℃,通入氧气至1.0mpa。于120℃,1.0mpa氧气压力,800rpm搅拌反应8.0h。反应完毕,冰水冷却至室温,向反应混合物中加入1.3115g(5.00mmol)三苯基膦(pph3),室温下搅拌30min还原生成的过氧化物。以丙酮为溶剂,将所得反应混合物定容至100ml。移取10ml所得溶液,以甲苯为内标,进行气相色谱分析;移取10ml所得溶液,以苯甲酸为内标,进行液相色谱分析。环己烷转化率2.43%,环己醇选择性9.0%,环己酮选择性18.7%,环己基过氧化氢选择性65.8%,己二酸选择性4.5%,戊二酸选择性2.0%。
实施例56(对比实验)
在100ml具有聚四氟乙烯内胆的不锈钢高压反应釜中,将0.0400gsi@p-cl-porp.co分散于16.8320g(200mmol)环己烷中,密封反应釜,搅拌升温到120℃,通入氧气至1.0mpa。于120℃,1.0mpa氧气压力,800rpm搅拌反应8.0h。反应完毕,冰水冷却至室温,向反应混合物中加入1.3115g(5.00mmol)三苯基膦(pph3),室温下搅拌30min还原生成的过氧化物。以丙酮为溶剂,将所得反应混合物定容至100ml。移取10ml所得溶液,以甲苯为内标,进行气相色谱分析;移取10ml所得溶液,以苯甲酸为内标,进行液相色谱分析。环己烷转化率3.61%,环己醇选择性33.3%,环己酮选择性36.7%,环己基过氧化氢选择性12.7%,己二酸选择性14.0%,戊二酸选择性3.3%。
实施例57(对比实验)
在100ml具有聚四氟乙烯内胆的不锈钢高压反应釜中,将0.0400gsi@p-cl-porp.cu分散于16.8320g(200mmol)环己烷中,密封反应釜,搅拌升温到120℃,通入氧气至1.0mpa。于120℃,1.0mpa氧气压力,800rpm搅拌反应8.0h。反应完毕,冰水冷却至室温,向反应混合物中加入1.3115g(5.00mmol)三苯基膦(pph3),室温下搅拌30min还原生成的过氧化物。以丙酮为溶剂,将所得反应混合物定容至100ml。移取10ml所得溶液,以甲苯为内标,进行气相色谱分析;移取10ml所得溶液,以苯甲酸为内标,进行液相色谱分析。环己烷转化率4.20%,环己醇选择性39.5%,环己酮选择性30.2%,环己基过氧化氢选择性15.0%,己二酸选择性11.2%,戊二酸选择性4.1%。
实施例58(放大实验)
在1.00l具有聚四氟乙烯内胆的不锈钢高压反应釜中,将0.4000gsi@p-cl-porp.co&zn分散于168.3200g(2.00mol)环己烷中,密封反应釜,搅拌升温到120℃,通入氧气至1.0mpa。于120℃,1.0mpa氧气压力,600rpm搅拌反应8.0h。反应完毕,冰水冷却至室温,向反应混合物中加入13.1145g(50.00mmol)三苯基膦(pph3),室温下搅拌30min还原生成的过氧化物。蒸馏,回收环己烷154.45g,转化率7.85%;减压精馏,得环己醇11.65g,选择性46.9%,环己酮9.96g,选择性48.0%。
- 还没有人留言评论。精彩留言会获得点赞!