用于氢化均苯四甲酸合成的催化剂的制作方法
103992330b)。目前,未见国内关于hpmda工业化的报道。目前,关于催化剂活性,选择性等催化剂技术指标均达到较高水平,产品hpma经分离纯化后可达到聚合级的纯度,但其hpma色度问题一定未能有效的解决,影响了产品hpma的后续应用。
技术实现要素:
7.本发明所要解决的技术问题之一是现有技术中存在的产品色度高的问题,提供一种新的氢化均苯四甲酸合成的催化剂,该催化剂具有氢化均苯四甲酸产品色度低的特点。
8.本发明所要解决的技术问题之二,是提供一种与上述技术问题之一相对应的催化剂的制备方法。
9.本发明所要解决的技术问题之三,是提供一种采用上述技术问题之一所述催化剂的氢化均苯四甲酸的合成方法。
10.为解决上述技术问题之一,本发明的技术方案如下:
11.用于氢化均苯四甲酸合成的催化剂,所述催化剂包括:载体,以及以每升催化剂计的负载于载体的如下组分:
12.贵金属,4.0-40g/l;
13.环糊精,0.4~4.0g/l;
14.所述载体为活性炭;所述贵金属包括选自由rh,ru,pd,pt和au所组成的物质组中的至少一种。
15.采用现有技术的催化剂,得到的氢化均苯四甲酸产品色度高,影响了产品后续应用。我们发现,由于本发明催化剂中加入环糊精,大幅度降低了氢化均苯四甲酸产品色度。
16.上述技术方案中,作为非限制性举例,所述催化剂中贵金属含量的例子可以但不限于是8.0g/l、12.0g/l、16.0g/l、20.0g/l、24.0g/l、28.0g/l、32.0g/l、36.0g/l等等。
17.上述技术方案中,作为非限制性举例,所述催化剂中环糊精含量是0.8g/l、1.2g/l、1.6g/l、2.0g/l、2.4g/l、2.8g/l、3.2g/l、3.6g/l等等。
18.上述技术方案中,所述环糊精优选包括由α-环糊精、由β-环糊精和由γ-环糊精所组成的物质组中的至少一种。
19.上述技术方案中,优选所述载体为椰壳炭。例如片状的椰壳炭,优选颗粒大小为10~30目。
20.上述技术方案中,优选载体比表面积为1000~2000m2/g,作为非限制性举例,载体比表面积可以是1050m2/g,1100m2/g,1150m2/g,1200m2/g,1250m2/g,1300m2/g,1350m2/g,1400m2/g,1450m2/g,1500m2/g,1550m2/g,1600m2/g,1650m2/g,1700m2/g,1750m2/g,1800m2/g,1850m2/g,1900m2/g,1950m2/g等等。
21.上述技术方案中,优选载体吸附总孔容为0.4~0.8cm3/g,作为非限制性举例,载体吸附总孔容可以是0.45cm3/g,0.50cm3/g,0.55cm3/g,0.60cm3/g,0.65cm3/g,0.70cm3/g,0.75cm3/g等等。
22.为解决上述技术问题之二,技术方案如下:
23.催化剂的制备方法,包括以下步骤:
24.(1)将贵金属盐溶液与活性炭混合,干燥得催化剂前驱体a;
25.(2)将催化剂前驱体a中化合态贵金属还原为零价,得到催化剂前驱体b;
26.(3)将催化剂前驱体b与环糊精水溶液混合,干燥后得催化剂;
27.上述技术方案中,步骤(1)中贵金属盐优选为盐酸盐、硝酸盐或醋酸盐。
28.上述技术方案中,步骤(1)中所述贵金属盐溶液中贵金属的浓度为4.0-40.0g/l,例如但不限于所述贵金属盐溶液中贵金属的浓度为8.0g/l、12.0g/l、16.0g/l、20.0g/l、24.0g/l、28.0g/l、32.0g/l、36.0g/l等等。
29.上述技术方案中,步骤(2)中的还原,可以是溶液还原或气体还原。
30.上述技术方案中,步骤(2)气体还原的还原剂优选h2气氛或h2和n2混合气。
31.上述技术方案中,步骤(2)气体还原的温度优选为200~500℃。例如但不限于250℃、300℃、350℃、400℃、450℃等等。
32.上述技术方案中,步骤(3)环糊精水溶液中环糊精的浓度为0.4-4.0g/l,例如但不限于浸渍液中环糊精的浓度为0.8g/l、1.2g/l、1.6g/l、2.0g/l、2.4g/l、2.8g/l、3.2g/l、3.6g/l等等。
33.为解决上述技术问题之三,本发明的技术方案如下:氢化均苯四甲酸合成方法,包括以均苯四甲酸和h2为原料,在上述技术问题之一的技术方案中任一项所述催化剂或按照上述技术问题之二的技术方案中任一项所述制备方法获得的催化剂的存在下,反应得到氢化均苯四甲酸。
34.本发明的技术关键是催化剂,而对于合成方法采用的具体工艺条件,本领域技术人员可以合理选择且均能取得可比的技术效果。但是:
35.上述技术方案中,反应温度优选为50~150℃,例如但不限于60℃、70℃、80℃、90℃、100℃、110℃、120℃、130℃、140℃等等。
36.上述技术方案中,反应中h2分压优选为5.0~10.0mpa,例如但不限于5.5mpa、6.0mpa、6.5mpa、7.0mpa、7.5mpa、8.0mpa、8.5mpa、9.0mpa、9.5mpa等等。
37.上述技术方案中,反应时间优选为1.0-10.0小时,例如但不限于2小时、3小时、4小时、5小时、6小时、7小时、8小时、9小时等等。
38.上述技术方案中,反应溶剂只要能溶解均苯四甲酸,并没有特别限定。例如但不限于水;甲醇,乙醇,1-丙醇,2-丙醇,1-丁醇,2-丁醇等醇类;二乙基醚,二异丁基醚,正丁基醚,thf等醚类;醋酸甲酯,醋酸乙酯等酯类;丙酮,甲乙酮等酮类。其中,优选是水,甲醇,乙醇,1-丙醇,2-丙醇,进一步优选是水。
39.本发明所述反应压力均为表压。
40.本发明均苯四甲酸转化率通过以下公式计算:
41.均苯四甲酸转化率(α)=(m
(pma,in)-m
(pma,out)
)/m
(pma,in)
×
100%
42.氢化均苯四甲酸选择性(s)=m
(hpma)/m(hpma)
/((m
(pma,in)-m
(pma,out)
)/m
(pma)
)
×
100%
43.氢化均苯四甲酸收率=α
×s×
100%
44.pma:均苯四甲酸;
45.hpma:氢化均苯四甲酸;
46.m
(pma,in)
:反应前投入pma质量;
47.m
(pma,out)
:反应后剩余pma质量;
48.m
(hpma)
:反应生成hpma质量;
49.m
(hpma)
:hpma的分子量;
50.m
(pma)
:pma的分子量;
51.hpma色度的测定采用分光光度法,选用agilent technologies cary 60 uv-vis仪器进行分析。
52.a)空白dmf的吸光度的测定:将n,n-二甲基甲酰胺注入5cm比色皿,在390nm波长处测得吸光度a0。
53.b)试样吸光度的测定:称取1.25g hpma,倒入装有15-20ml dmf的25ml容量瓶中,摇动溶解,然后将该容量瓶置于超声波容器中进行超声振荡溶解。待样品充分溶解后,用dmf定容至刻度并摇匀,过滤,得到透明澄清液体,将液体注入5cm比色皿,用390nm波长处用分光光度计测得其吸光度a1。
54.c)结果计算:
55.试样(5g/100ml dmf)色度x按式(1)计算:
56.x=150
×
(a1–
a0)
57.式中:
58.x——试样(5g/100ml dmf)色度,单位为铂-钴色号;
59.a1——试样溶液的吸光度;
60.a0——空白dmf的吸光度;
61.150——吸光度换算为铂-钴色号的常数。
具体实施方式
62.【实施例1】
63.(i)催化剂制备
64.(1)选用水合三氯化铑(rhcl3·
3h2o)与纯水混合,制得250.0ml含有5.0g rh的rhcl3水溶液为浸渍液1,其中rh浓度为20.0g/l。与250.0ml活性炭(片状椰壳炭,尺寸:20
±
2目,比表面:1500
±
50m2/g,孔容:0.6
±
0.03cm3/g)混合,在旋转蒸发仪上,60℃条件下浸渍2小时,减压蒸干溶剂后,将湿催化剂前驱体置于鼓风干燥箱中,120℃条件下干燥3小时,得催化剂前驱体a;
65.(2)将催化剂前驱体a置于管式气氛炉中,在含体积浓度10%h2的(h2和n2)混合气条件下,以10℃/min升温速率程序升温至300℃,恒温2小时,自然降温至室温25℃,得到催化剂前驱体b;
66.(3)选用α-环糊精(c
42h70o35
),配制250.0ml含有0.5gα-环糊精的水溶液为浸渍液2,其中α-环糊精的浓度为2.0g/l,与250.0ml催化剂前驱体b混合,在旋转蒸发仪上,在60℃条件下浸渍3小时,减压蒸干溶剂后,将湿催化剂置于鼓风干燥箱中,120℃条件下干燥3小时,得催化剂;
67.催化剂的制备条件和催化剂组成列于表1。
68.(ii)催化剂性能评价
69.用1l高压釜式反应器进行催化剂评价,具体反应和后处理步骤为:
70.a)在1000ml的高压釜中加入40.0g均苯四甲酸(c
10
h6o8,纯度:》98.0%),同时加入360g去离子水以及5.0g催化剂,向高压釜中通入氢气并将氢分压控制在7.5mpa,温度控制在100℃,反应3h。
71.b)然后冷却至室温(20
±
5℃)出料过滤,滤饼分离出rh/ac催化剂,该催化剂可循环使用;滤液减压蒸馏除水后,析出氢化均苯四甲酸(hpma),采用hplc进行定量分析,根据分析结果计算反应转化率、选择性、收率和纯度,采用分光光度计分析hpma色度。
72.为便于比较把催化剂主要制备条件列于表1,把催化剂主要评价条件列于表2,把反应产物分析列于表3。
73.【实施例2】
74.(i)催化剂制备
75.(1)选用水合三氯化铑(rhcl3·
3h2o)与纯水混合,制得250.0ml含有5.0g rh的rhcl3水溶液为浸渍液1,其中rh浓度为20.0g/l。与250.0ml活性炭(片状椰壳炭,尺寸:20
±
2目,比表面:1500
±
50m2/g,孔容:0.6
±
0.03cm3/g)混合,在旋转蒸发仪上,60℃条件下浸渍2小时,减压蒸干溶剂后,将湿催化剂前驱体置于鼓风干燥箱中,120℃条件下干燥3小时,得催化剂前驱体a;
76.(2)将催化剂前驱体a置于管式气氛炉中,在含体积浓度10%h2的(h2和n2)混合气条件下,以10℃/min升温速率程序升温至300℃,恒温2小时,自然降温至室温25℃,得到催化剂前驱体b;
77.(3)选用β-环糊精(c
42h70o35
),配制250.0ml含有0.5gβ-环糊精的水溶液为浸渍液2,其中β-环糊精的浓度为2.0g/l,与250.0ml催化剂前驱体b混合,在旋转蒸发仪上,在60℃条件下浸渍3小时,减压蒸干溶剂后,将湿催化剂置于鼓风干燥箱中,120℃条件下干燥3小时,得催化剂;
78.催化剂的制备条件和催化剂组成列于表1。
79.(ii)催化剂性能评价
80.用1l高压釜式反应器进行催化剂评价,具体反应和后处理步骤为:
81.a)在1000ml的高压釜中加入40.0g均苯四甲酸(c
10
h6o8,纯度:》98.0%),同时加入360g去离子水以及5.0g催化剂,向高压釜中通入氢气并将氢分压控制在7.5mpa,温度控制在100℃,反应3h。
82.b)然后冷却至室温(20
±
5℃)出料过滤,滤饼分离出rh/ac催化剂,该催化剂可循环使用;滤液减压蒸馏除水后,析出氢化均苯四甲酸(hpma),采用hplc进行定量分析,根据分析结果计算反应转化率、选择性、收率和纯度,采用分光光度计分析hpma色度。
83.为便于比较把催化剂主要制备条件列于表1,把催化剂主要评价条件列于表2,把反应产物分析列于表3。
84.【实施例3】
85.(i)催化剂制备
86.(1)选用水合三氯化铑(rhcl3·
3h2o)与纯水混合,制得250.0ml含有5.0g rh的rhcl3水溶液为浸渍液1,其中rh浓度为20.0g/l。与250.0ml活性炭(片状椰壳炭,尺寸:20
±
2目,比表面:1500
±
50m2/g,孔容:0.6
±
0.03cm3/g)混合,在旋转蒸发仪上,60℃条件下浸渍2小时,减压蒸干溶剂后,将湿催化剂前驱体置于鼓风干燥箱中,120℃条件下干燥3小时,得催化剂前驱体a;
87.(2)将催化剂前驱体a置于管式气氛炉中,在含体积浓度10%h2的(h2和n2)混合气条件下,以10℃/min升温速率程序升温至300℃,恒温2小时,自然降温至室温25℃,得到催
化剂前驱体b;
88.(3)选用γ-环糊精(c
48h80o40
),配制250.0ml含有0.5gγ-环糊精的水溶液为浸渍液2,其中γ-环糊精的浓度为2.0g/l,与250.0ml催化剂前驱体b混合,在旋转蒸发仪上,在60℃条件下浸渍3小时,减压蒸干溶剂后,将湿催化剂置于鼓风干燥箱中,120℃条件下干燥3小时,得催化剂;
89.催化剂的制备条件和催化剂组成列于表1。
90.(ii)催化剂性能评价
91.用1l高压釜式反应器进行催化剂评价,具体反应和后处理步骤为:
92.a)在1000ml的高压釜中加入40.0g均苯四甲酸(c
10
h6o8,纯度:》98.0%),同时加入360g去离子水以及5.0g催化剂,向高压釜中通入氢气并将氢分压控制在7.5mpa,温度控制在100℃,反应3h。
93.b)然后冷却至室温(20
±
5℃)出料过滤,滤饼分离出rh/ac催化剂,该催化剂可循环使用;滤液减压蒸馏除水后,析出氢化均苯四甲酸(hpma),采用hplc进行定量分析,根据分析结果计算反应转化率、选择性、收率和纯度,采用分光光度计分析hpma色度。
94.为便于比较把催化剂主要制备条件列于表1,把催化剂主要评价条件列于表2,把反应产物分析列于表3。
95.【实施例4】
96.(i)催化剂制备
97.(1)选用水合三氯化铑(rhcl3·
3h2o)与纯水混合,制得250.0ml含有1.0g rh的rhcl3水溶液为浸渍液1,其中rh浓度为4.0g/l。与250.0ml活性炭(片状椰壳炭,尺寸:10
±
2目,比表面:1000
±
50m2/g,孔容:0.4
±
0.02cm3/g)混合,在旋转蒸发仪上,60℃条件下浸渍2小时,减压蒸干溶剂后,将湿催化剂前驱体置于鼓风干燥箱中,120℃条件下干燥3小时,得催化剂前驱体a;
98.(2)将催化剂前驱体a置于管式气氛炉中,在含体积浓度10%h2的(h2和n2)混合气条件下,以10℃/min升温速率程序升温至200℃,恒温0.5小时,自然降温至室温25℃,得到催化剂前驱体b;
99.(3)选用β-环糊精(c
42h70o35
),配制250.0ml含有0.1gβ-环糊精的水溶液为浸渍液2,其中β-环糊精的浓度为0.4g/l,与250.0ml催化剂前驱体b混合,在旋转蒸发仪上,在60℃条件下浸渍3小时,减压蒸干溶剂后,将湿催化剂置于鼓风干燥箱中,120℃条件下干燥3小时,得催化剂;
100.催化剂的制备条件和催化剂组成列于表1。
101.(ii)催化剂性能评价
102.用1l高压釜式反应器进行催化剂评价,具体反应和后处理步骤为:
103.a)在1000ml的高压釜中加入10.0g均苯四甲酸(c
10
h6o8,纯度:》98.0%),同时加入190g去离子水以及5.0g催化剂,向高压釜中通入氢气并将氢分压控制在5.0mpa,温度控制在50℃,反应1h。
104.b)然后冷却至室温(20
±
5℃)出料过滤,滤饼分离出rh/ac催化剂,该催化剂可循环使用;滤液减压蒸馏除水后,析出氢化均苯四甲酸(hpma),采用hplc进行定量分析,根据分析结果计算反应转化率、选择性、收率和纯度,采用分光光度计分析hpma色度。
105.为便于比较把催化剂主要制备条件列于表1,把催化剂主要评价条件列于表2,把反应产物分析列于表3。
106.【实施例5】
107.(i)催化剂制备
108.(1)选用水合三氯化铑(rhcl3·
3h2o)与纯水混合,制得250.0ml含有10.0g rh的rhcl3水溶液为浸渍液1,其中rh浓度为40.0g/l。与250.0ml活性炭(片状椰壳炭,尺寸:30
±
2目,比表面:2000
±
50m2/g,孔容:0.8
±
0.04cm3/g)混合,在旋转蒸发仪上,60℃条件下浸渍2小时,减压蒸干溶剂后,将湿催化剂前驱体置于鼓风干燥箱中,120℃条件下干燥3小时,得催化剂前驱体a;
109.(2)将催化剂前驱体a置于管式气氛炉中,在含体积浓度10%h2的(h2和n2)混合气条件下,以10℃/min升温速率程序升温至500℃,恒温5小时,自然降温至室温25℃,得到催化剂前驱体b;
110.(3)选用β-环糊精(c
42h70o35
),配制250.0ml含有1.0gβ-环糊精的水溶液为浸渍液2,其中β-环糊精的浓度为4.0g/l,与250.0ml催化剂前驱体b混合,在旋转蒸发仪上,在60℃条件下浸渍3小时,减压蒸干溶剂后,将湿催化剂置于鼓风干燥箱中,120℃条件下干燥3小时,得催化剂;
111.催化剂的制备条件和催化剂组成列于表1。
112.(ii)催化剂性能评价
113.用1l高压釜式反应器进行催化剂评价,具体反应和后处理步骤为:
114.a)在1000ml的高压釜中加入100.0g均苯四甲酸(c
10
h6o8,纯度:》98.0%),同时加入400g去离子水以及5.0g催化剂,向高压釜中通入氢气并将氢分压控制在10.0mpa,温度控制在150℃,反应10h。
115.b)然后冷却至室温(20
±
5℃)出料过滤,滤饼分离出rh/ac催化剂,该催化剂可循环使用;滤液减压蒸馏除水后,析出氢化均苯四甲酸(hpma),采用hplc进行定量分析,根据分析结果计算反应转化率、选择性、收率和纯度,采用分光光度计分析hpma色度。
116.为便于比较把催化剂主要制备条件列于表1,把催化剂主要评价条件列于表2,把反应产物分析列于表3。
117.【实施例6】
118.(i)催化剂制备
119.(1)选用三氯化钌三水合物(rucl3·
3h2o)与纯水混合,制得250.0ml含有5.0g ru的rucl3水溶液为浸渍液1,其中ru浓度为20.0g/l。与250.0ml活性炭(片状椰壳炭,尺寸:20
±
2目,比表面:1500
±
50m2/g,孔容:0.6
±
0.03cm3/g)混合,在旋转蒸发仪上,60℃条件下浸渍2小时,减压蒸干溶剂后,将湿催化剂前驱体置于鼓风干燥箱中,120℃条件下干燥3小时,得催化剂前驱体a;
120.(2)将催化剂前驱体a置于管式气氛炉中,在含体积浓度10%h2的(h2和n2)混合气条件下,以10℃/min升温速率程序升温至300℃,恒温2小时,自然降温至室温25℃,得到催化剂前驱体b;
121.(3)选用α-环糊精(c
42h70o35
),配制250.0ml含有0.5gα-环糊精的水溶液为浸渍液2,其中α-环糊精的浓度为2.0g/l,与250.0ml催化剂前驱体b混合,在旋转蒸发仪上,在60℃
条件下浸渍3小时,减压蒸干溶剂后,将湿催化剂置于鼓风干燥箱中,120℃条件下干燥3小时,得催化剂;
122.催化剂的制备条件和催化剂组成列于表1。
123.(ii)催化剂性能评价
124.用1l高压釜式反应器进行催化剂评价,具体反应和后处理步骤为:
125.a)在1000ml的高压釜中加入40.0g均苯四甲酸(c
10
h6o8,纯度:》98.0%),同时加入360g去离子水以及5.0g催化剂,向高压釜中通入氢气并将氢分压控制在7.5mpa,温度控制在100℃,反应3h。
126.b)然后冷却至室温(20
±
5℃)出料过滤,滤饼分离出ru/ac催化剂,该催化剂可循环使用;滤液减压蒸馏除水后,析出氢化均苯四甲酸(hpma),采用hplc进行定量分析,根据分析结果计算反应转化率、选择性、收率和纯度,采用分光光度计分析hpma色度。
127.为便于比较把催化剂主要制备条件列于表1,把催化剂主要评价条件列于表2,把反应产物分析列于表3。
128.【实施例7】
129.(i)催化剂制备
130.(1)选用三氯化钌三水合物(rucl3·
3h2o)与纯水混合,制得250.0ml含有5.0g ru的rucl3水溶液为浸渍液1,其中ru浓度为20.0g/l。与250.0ml活性炭(片状椰壳炭,尺寸:20
±
2目,比表面:1500
±
50m2/g,孔容:0.6
±
0.03cm3/g)混合,在旋转蒸发仪上,60℃条件下浸渍2小时,减压蒸干溶剂后,将湿催化剂前驱体置于鼓风干燥箱中,120℃条件下干燥3小时,得催化剂前驱体a;
131.(2)将催化剂前驱体a置于管式气氛炉中,在含体积浓度10%h2的(h2和n2)混合气条件下,以10℃/min升温速率程序升温至300℃,恒温2小时,自然降温至室温25℃,得到催化剂前驱体b;
132.(3)选用β-环糊精(c
42h70o35
),配制250.0ml含有0.5gβ-环糊精的水溶液为浸渍液2,其中β-环糊精的浓度为2.0g/l,与250.0ml催化剂前驱体b混合,在旋转蒸发仪上,在60℃条件下浸渍3小时,减压蒸干溶剂后,将湿催化剂置于鼓风干燥箱中,120℃条件下干燥3小时,得催化剂;
133.催化剂的制备条件和催化剂组成列于表1。
134.(ii)催化剂性能评价
135.用1l高压釜式反应器进行催化剂评价,具体反应和后处理步骤为:
136.a)在1000ml的高压釜中加入40.0g均苯四甲酸(c
10
h6o8,纯度:》98.0%),同时加入360g去离子水以及5.0g催化剂,向高压釜中通入氢气并将氢分压控制在7.5mpa,温度控制在100℃,反应3h。
137.b)然后冷却至室温(20
±
5℃)出料过滤,滤饼分离出ru/ac催化剂,该催化剂可循环使用;滤液减压蒸馏除水后,析出氢化均苯四甲酸(hpma),采用hplc进行定量分析,根据分析结果计算反应转化率、选择性、收率和纯度,采用分光光度计分析hpma色度。
138.为便于比较把催化剂主要制备条件列于表1,把催化剂主要评价条件列于表2,把反应产物分析列于表3。
139.【实施例8】
140.(i)催化剂制备
141.(1)选用三氯化钌三水合物(rucl3·
3h2o)与纯水混合,制得250.0ml含有5.0g ru的rucl3水溶液为浸渍液1,其中ru浓度为20.0g/l。与250.0ml活性炭(片状椰壳炭,尺寸:20
±
2目,比表面:1500
±
50m2/g,孔容:0.6
±
0.03cm3/g)混合,在旋转蒸发仪上,60℃条件下浸渍2小时,减压蒸干溶剂后,将湿催化剂前驱体置于鼓风干燥箱中,120℃条件下干燥3小时,得催化剂前驱体a;
142.(2)将催化剂前驱体a置于管式气氛炉中,在含体积浓度10%h2的(h2和n2)混合气条件下,以10℃/min升温速率程序升温至300℃,恒温2小时,自然降温至室温25℃,得到催化剂前驱体b;
143.(3)选用γ-环糊精(c
48h80o40
),配制250.0ml含有0.5gγ-环糊精的水溶液为浸渍液2,其中γ-环糊精的浓度为2.0g/l,与250.0ml催化剂前驱体b混合,在旋转蒸发仪上,在60℃条件下浸渍3小时,减压蒸干溶剂α后,将湿催化剂置于鼓风干燥箱中,120℃条件下干燥3小时,得催化剂;
144.催化剂的制备条件和催化剂组成列于表1。
145.(ii)催化剂性能评价
146.用1l高压釜式反应器进行催化剂评价,具体反应和后处理步骤为:
147.a)在1000ml的高压釜中加入40.0g均苯四甲酸(c
10
h6o8,纯度:》98.0%),同时加入360g去离子水以及5.0g催化剂,向高压釜中通入氢气并将氢分压控制在7.5mpa,温度控制在100℃,反应3h。
148.b)然后冷却至室温(20
±
5℃)出料过滤,滤饼分离出ru/ac催化剂,该催化剂可循环使用;滤液减压蒸馏除水后,析出氢化均苯四甲酸(hpma),采用hplc进行定量分析,根据分析结果计算反应转化率、选择性、收率和纯度,采用分光光度计分析hpma色度。
149.为便于比较把催化剂主要制备条件列于表1,把催化剂主要评价条件列于表2,把反应产物分析列于表3。
150.【实施例9】
151.(i)催化剂制备
152.(1)选用氯钯酸(h2pdcl4)与纯水混合,制得250.0ml含有5.0g pd的h2pdcl4水溶液为浸渍液1,其中pd浓度为20.0g/l。与250.0ml活性炭(片状椰壳炭,尺寸:20
±
2目,比表面:1500
±
50m2/g,孔容:0.6
±
0.03cm3/g)混合,在旋转蒸发仪上,60℃条件下浸渍2小时,减压蒸干溶剂后,将湿催化剂前驱体置于鼓风干燥箱中,120℃条件下干燥3小时,得催化剂前驱体a;
153.(2)将催化剂前驱体a置于管式气氛炉中,在含体积浓度10%h2的(h2和n2)混合气条件下,以10℃/min升温速率程序升温至300℃,恒温2小时,自然降温至室温25℃,得到催化剂前驱体b;
154.(3)选用β-环糊精(c
42h70o35
),配制250.0ml含有0.5gβ-环糊精的水溶液为浸渍液2,其中β-环糊精的浓度为2.0g/l,与250.0ml催化剂前驱体b混合,在旋转蒸发仪上,在60℃条件下浸渍3小时,减压蒸干溶剂后,将湿催化剂置于鼓风干燥箱中,120℃条件下干燥3小时,得催化剂;
155.催化剂的制备条件和催化剂组成列于表1。
目,比表面:1500
±
50m2/g,孔容:0.6
±
0.03cm3/g)混合,在旋转蒸发仪上,60℃条件下浸渍2小时,减压蒸干溶剂后,将湿催化剂前驱体置于鼓风干燥箱中,120℃条件下干燥3小时,得催化剂前驱体a;
175.(2)将催化剂前驱体a置于管式气氛炉中,在含体积浓度10%h2的(h2和n2)混合气条件下,以10℃/min升温速率程序升温至300℃,恒温2小时,自然降温至室温25℃,得到催化剂成品b;
176.催化剂的制备条件和催化剂组成列于表1。
177.(ii)催化剂性能评价
178.用1l高压釜式反应器进行催化剂评价,具体反应和后处理步骤为:
179.a)在1000ml的高压釜中加入40.0g均苯四甲酸(c
10
h6o8,纯度:》98.0%),同时加入360g去离子水以及5.0g ru/ac贵金属催化剂,向高压釜中通入氢气并将氢分压控制在7.5mpa,温度控制在100℃,反应3h。
180.b)然后冷却至室温(20
±
5℃)出料过滤,滤饼分离出rh/ac催化剂,该催化剂可循环使用;滤液减压蒸馏除水后,析出氢化均苯四甲酸(hpma),采用hplc进行定量分析,根据分析结果计算反应转化率、选择性、收率和纯度,采用分光光度计分析hpma色度。
181.为便于比较把催化剂主要制备条件列于表1,把催化剂主要评价条件列于表2,把反应产物分析列于表3。
182.应当注意的是,以上所述的实施例仅用于解释本发明,并不构成对本发明的任何限制。通过参照典型实施例对本发明进行了描述,但应当理解为其中所用的词语为描述性和解释性词汇,而不是限定性词汇。可以按规定在本发明权利要求的范围内对本发明作出修改,以及在不背离本发明的范围和精神内对本发明进行修订。尽管其中描述的本发明涉及特定的方法、材料和实施例,但是并不意味着本发明限于其中公开的特定例,相反,本发明可扩展至其他所有具有相同功能的方法和应用。
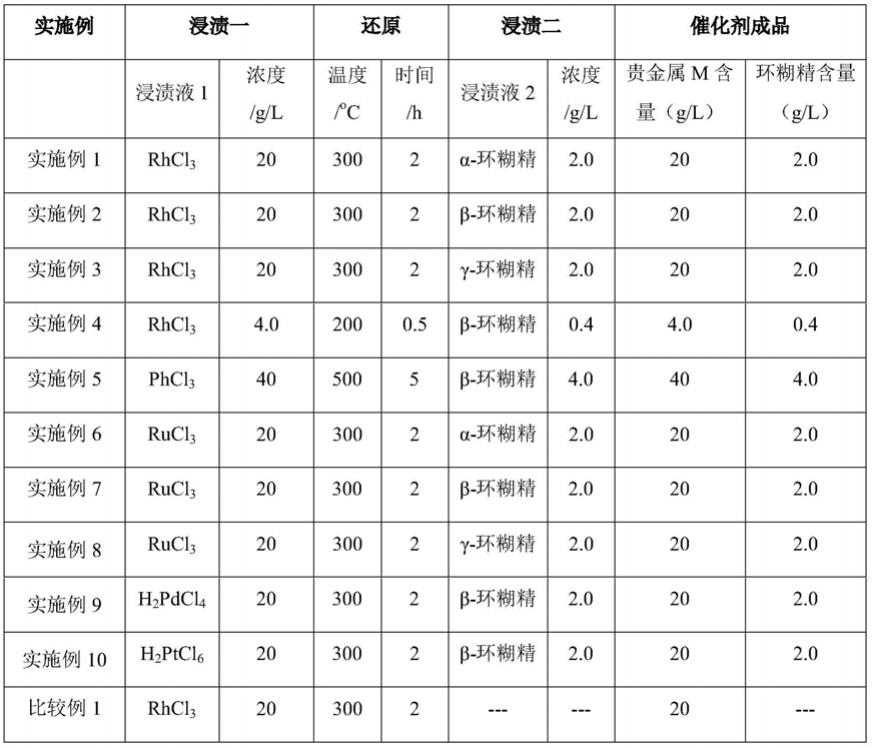
183.表1催化剂制备与表征
[0184][0185]
表2
[0186] h2分压反应温度反应时间反应溶剂原料溶液浓度原料与催化剂之比 (mpa)(℃)(h) (%) 实施例17.51003h2o108:1实施例27.51003h2o108:1实施例37.51003h2o108:1实施例45.0501h2o52:1实施例510.015010h2o2020:1实施例67.51003h2o108:1实施例77.51003h2o108:1实施例87.51003h2o108:1实施例97.51003h2o108:1实施例107.51003h2o108:1比较例17.51003h2o108:1
[0187]
表3
[0188] 转化率选择性收率纯度色度 (%)(%(%)(%) 实施例199.699.198.799.128实施例299.899.599.399.515实施例399.799.399.099.324
实施例498.198.896.998.932实施例599.098.597.598.829实施例699.198.998.099.138实施例799.099.298.299.234实施例899.398.898.198.826实施例998.698.797.398.428实施例1099.498.497.899.019比较例195.996.8.92.895.5120
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1