脱除芳烃中氮化物的吸附剂及其制备方法和应用与流程
1.本发明涉及一种脱除芳烃中氮化物的吸附剂及其制备方法。
背景技术:
2.芳烃,如苯、甲苯、二甲苯、乙苯、异丙苯等是非常重要的基本有机化工原料。苯和乙烯在分子筛催化剂存在时可以发生烷基化反应生成乙苯,乙苯在催化剂存在下直接脱氢或氧化脱氢可生成苯乙烯,作为聚苯乙烯和abs树脂的单体使用;苯和丙烯在分子筛催化剂存在时发生烷基化反应生成异丙苯,异丙苯进一步氧化生成过氧化氢异丙苯,过氧化氢异丙苯既可以在酸作用下分解生产苯酚、丙酮,也可以在含钛二氧化硅催化剂存在下选择氧化丙烯生产环氧丙烷;甲苯可以在分子筛催化剂存在下发生歧化或择形歧化反应生成对二甲苯。上述反应均需在催化剂上完成,而这些催化剂对原料中的杂质,特别是氮化物非常敏感,氮化物呈碱性,微量氮化物容易吸附到催化剂活性中心上造成催化剂失活,影响反应的正常进行和工业装置的稳定运行。芳烃中氮化物主要为吡啶、喹啉、n-甲酰基
-ꢀ
吗啉和n-甲基-吡咯烷酮等,这些氮化物在分子大小及极性上具有一定的相似性,因此,这些极性杂质和芳烃间存在着严重的竞争吸附作用,影响杂质的脱除效果。
3.cn1461290a公开了一种从芳族原料中脱除极性杂质的方法,该方法将含极性化合物的原料与一种吸附剂接触,所述吸附剂包括一种孔和/或表面凹坑的和横截面尺寸大于5.6 埃的分子筛,得到处理后的原料进入烷基化反应单元。
4.cn107915567a公开了一种从含极性化合物的芳族原料中脱除极性化合物的方法,该方法通过采用含极性化合物的芳族原料进入精馏塔,塔釜得到第一物流,塔顶得到第二物流;所述第一物流进入吸附区,采用酸性白土或沸石为吸附剂,与吸附剂接触后,得到基本不含极性化合物的芳族原料。
5.cn107970781a公开了一种用于烯烃净化的分子筛陶瓷膜材料及其制备与应用,分子筛陶瓷膜材料中,陶瓷材料表面负载的分子筛颗粒的粒径为0.1-3μm,分子筛层的厚度为 3-5μm;制备时,依次经陶瓷材料预处理、分子筛晶种预涂覆、密闭晶化即可;分子筛陶瓷膜材料用于将气态烯烃流中的极性含氧化合物脱除至1ppm以下。
6.但是,现有技术在脱除氮化物的过程中仍存在吸附剂吸附容量低,脱除精度不够,固废排放量大的问题。
技术实现要素:
7.本发明所要解决的技术问题是现有技术中吸附剂吸附容量低,脱除精度不够的问题,提供一种脱除芳烃中氮化物的吸附剂及其制备方法。
8.为解决上述技术问题,本发明第一方面在于提供一种脱除芳烃中氮化物的吸附剂,所述吸附剂以重量份数计,包括:a)30~70份分子筛;b)10~40份的拟薄水铝石;c)10~40 份的氧化铝;d)0.5~10份的稀土元素;吸附剂强弱酸比≥0.25,优选≥0.50,更优选为0.6~0.8。
9.上述技术方案中,吸附剂强弱酸比为吸附剂强酸酸量与弱酸酸量之比。温度小于300℃酸中心为弱酸中心,温度大于300℃酸中心为强酸中心。上述技术方案中,分子筛选自x 型分子筛、y型分子筛一种或多种,优选13x和nay中的一种或两种。
10.上述技术方案中,以重量份数计分子筛含量优选35~50份;拟薄水铝石含量优选25~35 份;氧化铝含量优选25-35份;稀土元素含量优选为5~8份。
11.上述技术方案中,所述拟薄水铝石的比表面积300~400m2/g,孔容0.3~0.4cm3/g,弱酸酸量为0.20~0.50mmol/g,孔径为4.0~5.0nm。
12.上述技术方案中,所述氧化铝的比表面积200~300m2/g,孔容0.5~0.8cm3/g,孔径为 6.0~8.0nm,弱酸酸量为0.01~0.05mmol/g。
13.上述技术方案中,稀土元素选自la、ce、pr或nd中的至少一种。
14.本发明第二方面在于提供一种上述吸附剂的制备方法,包括以下步骤:
15.(1)在共沉淀反应器并流加入偏铝酸钠溶液和硝酸溶液进行共沉淀反应,得到拟薄水铝石;
16.(2)在步骤(1)所得到的拟薄水铝石中加入0.5%~1.5%聚丙烯酸溶液混合均匀,固液质量比为1:0.5~1:1.5,再经干燥、焙烧,制得氧化铝;
17.(3)将分子筛与来自步骤(1)的拟薄水铝石和来自步骤(2)的氧化铝捏合成型,干燥、焙烧,得到吸附剂前体;
18.(4)将步骤(3)得到的吸附剂前体等体积浸渍稀土元素盐溶液,干燥、焙烧,得到吸附剂。
19.上述技术方案中,步骤(1)所述共沉淀反应的反应温度为40~90℃,ph=6~8,反应时间为0.5~3小时。偏铝酸钠溶液浓度为0.5mol/l~1.5mol/l。偏铝酸钠溶液以例如 5ml/min~40ml/min速度加入和硝酸溶液反应,硝酸溶液的加入量使得反应的ph=6~8。反应得沉淀物,将沉淀物洗涤干燥,得到拟薄水铝石。
20.上述技术方案中,步骤(2)所述干燥温度为80~150℃;焙烧温度为400~600℃。
21.上述技术方案中,步骤(3)中捏合过程优选加入本领域任何可用的胶溶剂和助挤剂。所述干燥的温度为50~150℃,所述焙烧为300~600℃焙烧2~10小时。各物质的投料量,以重量份数计分子筛30~70份,优选35~50份;拟薄水铝石10~40份,优选25~35份;氧化铝10~40份,优选25-35份。
22.上述技术方案中,步骤(4)中稀土元素盐溶液的质量浓度为2%~13%。
23.上述技术方案中,步骤(4)中所述焙烧为400~600℃焙烧2~10小时。
24.本发明第三方面在于提供一种所述吸附剂在脱除芳烃中的氮化物中的应用。
25.上述技术方案中,所述氮化物包括吡啶、喹啉、n-甲酰基-吗啉和n-甲基-吡咯烷酮等中的一种或多种。
26.本发明所述吸附剂,由于采用分子筛、拟薄水铝石、氧化铝调和的方案对吸附剂的极性和酸性进行调和,使吸附剂具有适合的酸性和极性,加入稀土元素改善了了吸附剂电荷配位,稀土金属离子的d空轨道和f空轨道与碱性氮化合物p-π轨道之间将产生强的吸附作用,提高了与氮化物的接触能力,提高吸附容量和脱除精度。本发明取得了较好的技术效果。
附图说明
[0027][0028]
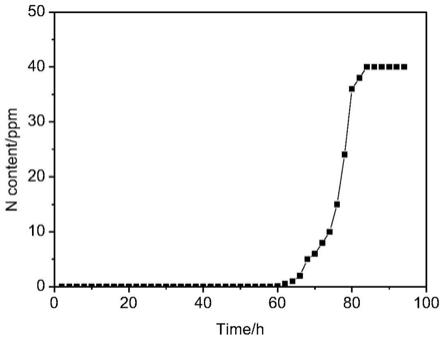
图1实施例1所制备吸附剂的穿透曲线。
具体实施方式
[0029]
下面通过具体实施例对本发明作进一步的阐述。但应当理解具体实施方式仅为更好地阐述发明内容,并不构成对保护范围的限制。
[0030]
样品的酸中心在天津市鹏翔科技有限公司的px200a型程序升温热脱附装置上进行 nh
3-tpd测试,氨吸附温度为100℃,载气为he,流速30ml
·
min-1
,升温速率为10℃
·
min-1
。温度小于300℃酸中心为弱酸中心,温度大于300℃为强酸中心。总酸量通过微量天平称取得到。积分拟合分别得到强酸中心酸量和弱酸中心酸量峰面积,通多计算分别得到强酸中心和弱酸中心的量。
[0031]
采用美国asap2600型表面分析仪测定样品的吸附曲线,根据吸附曲线采用bet法计算样品比表面积,用单点法计算总孔容。
[0032]
吸附剂穿透曲线的测定:将吸附剂装入固定床反应器,在180℃活化3小时,通入含 200ppm吡啶苯原料,室温,常压,空速5h-1
,用硫氮分析仪测定出口原料中氮含量,当出口原料氮含量超过1ppm则视为吸附剂穿透,得到穿透曲线。根据穿透曲线计算吸附剂的穿透吸附量。
[0033]
【实施例1】
[0034]
在共沉淀反应器中加入400ml蒸馏水,以35ml/min速度加入浓度为1.0mol/l的偏铝酸钠溶液和硝酸溶液反应,在60℃,ph=7条件下,反应2小时得沉淀物,将沉淀物洗涤干燥,得到拟薄水铝石。比表面积330m2/g,孔容0.36cm3/g。弱酸酸量为0.45mmol/g。
[0035]
将100克上述拟薄水铝石加入50克0.5%聚丙烯酸溶液混合均匀,在120℃,干燥,在550℃焙烧,得到氧化铝。比表面积250m2/g,孔容0.55cm3/g。弱酸酸量为0.02mmol/g。
[0036]
将50克13x、25克拟薄水铝石、25克氧化铝及5克田菁粉打粉,混合均匀后,加入 60克hno3(5%)捏合,捏合后,挤条成型,120℃干燥12小时。挤条成型样品在300℃焙烧5小时,得到吸附剂前体。用10%ce(no3)3溶液等体积浸渍吸附剂前体(含稀土元素 5.4克),在80℃烘干12小时,再在500℃焙烧5小时,即得吸附剂。吸附剂强弱酸比为 0.75。
[0037]
吸附剂吡啶吸附量的测定:将5g吸附剂装入固定床反应器,在180℃活化3小时,通入含200ppm吡啶苯原料,室温,常压,空速5h-1
,用硫氮分析仪测定出口原料中氮含量,当出口原料氮含量超过1ppm则视为吸附剂穿透,通过穿透曲线计算吸附剂的吡啶穿透吸附量为2.4%。穿透曲线如图1所示。
[0038]
【实施例2】
[0039]
在共沉淀反应器中加入200ml蒸馏水,以25ml/min速度加入浓度为1.5mol/l的偏铝酸钠溶液和硝酸溶液反应,在80℃,ph=6,反应0.5小时得沉淀物,将沉淀物洗涤干燥,得到拟薄水铝石。比表面积315m2/g,孔容0.32cm3/g。弱酸酸量为0.38mmol/g。将100克上述拟薄水铝石加入100克1.0%聚丙烯酸溶液混合均匀,在80℃,干燥,在450℃焙烧,得到氧化铝。比表面积238m2/g,孔容0.45cm3/g。弱酸酸量为0.01mmol/g。
[0040]
将35克13x、30克拟薄水铝石、35克氧化铝及5克田菁粉打粉,混合均匀后,加入 60
克hno3(5%)捏合,捏合后,挤条成型,120℃干燥12小时。挤条成型样品在600℃焙烧2小时,得到吸附剂前体。用10%ce(no3)3溶液等体积浸渍吸附剂前体(含稀土元素 5.4克),在80℃烘干12小时,再在600℃焙烧3小时,即得吸附剂。吸附剂强弱酸比为0.79。
[0041]
吸附剂吡啶吸附量的测定:将5g吸附剂装入固定床反应器,在180℃活化3小时,通入含200ppm吡啶苯原料,室温,常压,空速5h-1
,用硫氮分析仪测定出口原料中氮含量含量,当出口原料氮含量超过1ppm则视为吸附剂穿透,通过穿透曲线计算吸附剂的吡啶穿透吸附量为3.1%。
[0042]
【实施例3】
[0043]
在共沉淀反应器中加入400ml蒸馏水,以30ml/min速度加入浓度为1.5mol/l的偏铝酸钠溶液和硝酸溶液反应,在50℃,ph=8,反应3小时得沉淀物,将沉淀物洗涤干燥,得到拟薄水铝石。比表面积350m2/g,孔容0.38cm3/g。弱酸酸量为0.0.47mmol/g。将100 克上述拟薄水铝石加入150克1.5%聚丙烯酸溶液混合均匀,在150℃,干燥,在400℃焙烧,得到氧化铝。比表面积280m2/g,孔容0.65cm3/g。弱酸酸量为0.02mmol/g。
[0044]
将60克nay、40克拟薄水铝石、30克氧化铝及5克田菁粉打粉,混合均匀后,加入 60克hno3(5%)捏合,捏合后,挤条成型,120℃干燥12小时。挤条成型样品在300℃焙烧5小时,得到吸附剂前体。用10%ce(no3)3溶液等体积浸渍吸附剂前体(含稀土元素 7.0克),在80℃烘干12小时,再在500℃焙烧5小时,即得吸附剂。吸附剂强弱酸比为 0.69。
[0045]
吸附剂吡啶吸附量的测定:将5g吸附剂装入固定床反应器,在180℃活化3小时,通入含200ppm吡啶苯原料,室温,常压,空速5h-1
,用硫氮分析仪测定出口原料中吡啶含量,当出口原料吡啶超过1ppm则视为吸附剂穿透,通过穿透曲线计算吸附剂的吡啶穿透吸附量为2.0%。
[0046]
【实施例4】
[0047]
在共沉淀反应器中加入400ml蒸馏水,以35ml/min速度加入浓度为1.0mol/l的偏铝酸钠溶液和硝酸溶液反应,在60℃,ph=7,反应2小时得沉淀物,将沉淀物洗涤干燥,得到拟薄水铝石。比表面积330m2/g,孔容0.36cm3/g。弱酸酸量为0.45mmol/g。
[0048]
将100克上述拟薄水铝石加入50克0.5%聚丙烯酸溶液混合均匀,在120℃,干燥,在550℃焙烧,得到氧化铝。比表面积250m2/g,孔容0.55cm3/g。弱酸酸量为0.02mmol/g。
[0049]
将70克13x、35克拟薄水铝石、35克氧化铝及5克田菁粉打粉,混合均匀后,加入 60克hno3(5%)捏合,捏合后,挤条成型,120℃干燥12小时。挤条成型样品在300℃焙烧5小时,得到吸附剂前体。用10%ce(no3)3溶液等体积浸渍吸附剂前体(含稀土元素7.6克),在80℃烘干12小时,再在500℃焙烧5小时,即得吸附剂。吸附剂强弱酸比为 0.80。
[0050]
吸附剂吡啶吸附量的测定:将5g吸附剂装入固定床反应器,在180℃活化3小时,通入含200ppm吡啶苯原料,室温,常压,空速5h-1
,用硫氮分析仪测定出口原料中氮含量,当出口原料氮含量超过1ppm则视为吸附剂穿透,通过穿透曲线计算吸附剂的吡啶穿透吸附量为3.3%。
[0051]
【比较例1】
[0052]
在共沉淀反应器中加入400ml蒸馏水,以35ml/min速度加入浓度为1.0mol/l的偏铝酸钠溶液和硝酸溶液反应,在60℃,ph=7,反应2小时得沉淀物,将沉淀物洗涤干燥,得到拟薄水铝石。比表面积330m2/g,孔容0.36cm3/g。弱酸酸量为0.45mmol/g。
[0053]
将70克13x、35克拟薄水铝石及5克田菁粉打粉,混合均匀后,加入60克hno3(5%) 捏合,捏合后,挤条成型,120℃干燥12小时。挤条成型样品在300℃焙烧5小时,得到吸附剂前体。用1%ce(no3)3溶液等体积浸渍吸附剂前体,在80℃烘干12小时,再在500℃焙烧5小时,即得吸附剂。吸附剂强弱酸比为0.15。
[0054]
吸附剂吡啶吸附量的测定:将5g吸附剂装入固定床反应器,在180℃活化3小时,通入含200ppm吡啶苯原料,室温,常压,空速5h-1
,用硫氮分析仪测定出口原料中氮含量,当出口原料氮含量超过1ppm则视为吸附剂穿透,通过穿透曲线计算吸附剂的吡啶穿透吸附量为1.3%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1