一种加氢催化剂及其制备方法和应用以及环己基甲酸的制备方法与流程
1.本发明涉及一种加氢催化剂及其制备方法和应用,本发明还涉及采用所述加氢催化剂的环己基甲酸的制备方法。
背景技术:
2.环己基甲酸(即,)及其衍生物是重要的有机化工原料和药物的中间体,在药物合成及新材料研发中具有广泛的应用价值。环己基甲酸主要用于光引发剂184(即,1-羧基环已基苯基甲酮)的合成,还可用作硫化橡胶的增溶剂、石油的澄清剂和医药中间体,用于药物抗孕392和治疗血吸虫病药物吡喹酮的合成。
3.环己基甲酸可以通过将苯甲酸进行加氢制得。苯甲酸加氢制环己基甲酸的催化剂以pd/c或改进的pd/c催化剂为主,加氢工艺主要采用釜式加氢工艺。
4.cn101092349a公开了一种苯甲酸的加氢方法,在pd/c催化剂和ru/c助剂的存在下,熔融的苯甲酸与氢气在反应器中进行加氢反应,反应后混合物经过旋液分离和离心分离,含有高浓度催化剂和助剂的浊液循环回反应器系统,分离出的清液进入蒸发器进行进一步分离,其中,蒸发器分离出的催化剂和助剂完全返回反应器,其中,加氢反应温度为120-180℃。该方法的不足之处在于:首先,熔融的苯甲酸反应活性低,工艺条件苛刻,并且贵金属钯价格昂贵;其次,采用釜式反应工艺进行苯甲酸加氢反应时,工艺较为复杂,并且产物与催化剂长时间接触,使二次反应增多,反应选择性下降,还易导致催化剂中毒,缩短催化剂的使用寿命;另外,由于加氢催化剂通常为粉末状,与反应原料和产物的分离难度高,在过滤分离和催化剂再生过程中的损失较大,导致贵金属催化剂的单耗较高。
5.综上所述,亟需开发新型苯甲酸加氢催化剂及苯甲酸加氢工艺。
技术实现要素:
6.本发明的目的在于克服现有技术的不足,提供一种加氢催化剂及其制备方法,该加氢催化剂用作苯甲酸加氢制备环己基甲酸反应的催化剂时,能在固定床反应器中于温和的条件下进行加氢,并具有较高的催化活性。
7.根据本发明的第一个方面,本发明提供了一种加氢催化剂,该加氢催化剂含有载体以及负载在所述载体上的活性组分、助剂组分和碱金属元素,所述活性组分为钌,所述助剂组分为镍、铁和钴中的一种或两种以上。
8.根据本发明的第二个方面,本发明提供了一种加氢催化剂的制备方法,该方法包括以下步骤:
9.(1)将载体与含有碱金属化合物前身物的溶液接触,得到改性载体;
10.(2)将所述改性载体与含有活性组分前身物和助剂组分前身物的溶液接触,得到
负载有活性组分前身物和助剂组分前身物的负载载体,脱除所述负载载体中的至少部分挥发性组分后进行焙烧,得到加氢催化剂前体,所述焙烧在不高于300℃的温度下进行,所述活性组分前身物中的活性组分为钌,所述助剂组分前身物中的助剂组分为镍、铁和钴中的一种或两种以上;
11.(3)在还原反应条件下,将所述加氢催化剂前体与还原剂接触,得到所述加氢催化剂。
12.根据本发明的第三个方面,本发明提供了一种由本发明第二个方面所述的方法制备的加氢催化剂。
13.根据本发明的第四个方面,本发明提供了本发明第一个方面或者第三个方面所述的加氢催化剂作为苯甲酸加氢反应的催化剂的应用。
14.根据本发明的第五个方面,本发明提供了一种环己基甲酸的制备方法,该方法包括在加氢反应条件下,在加氢催化剂存在下,将苯甲酸与氢气接触,其中,所述加氢催化剂为本发明第一个方面或者第三个方面所述的加氢催化剂。
15.根据本发明的加氢催化剂即便在低温下也具有较高的催化活性,作为由苯甲酸制备环己基甲酸的加氢反应的催化剂,能在较为温和的反应条件下进行反应,并且能获得提高的催化活性。根据本发明的加氢催化剂适合用于在固定床反应器中进行苯甲酸加氢反应,能够实现连续便捷的操作,避免采用釜式反应器所必须的催化剂与反应物的分离操作,减少催化剂的损耗。根据本发明的环己基甲酸的制备方法可实现装置的连续稳定运行,满足工业化规模的操作要求。
具体实施方式
16.在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
17.根据本发明的第一个方面,本发明提供了一种加氢催化剂,该加氢催化剂含有载体以及负载在所述载体上的活性组分、助剂组分和碱金属元素。
18.根据本发明的加氢催化剂,所述活性组分为钌。根据本发明的加氢催化剂,以加氢催化剂的总量为基准,所述活性组分的含量优选为0.3-3重量%,例如可以为:0.3、0.4、0.5、0.6、0.7、0.8、0.9、1、1.1、1.2、1.3、1.4、1.5、1.6、1.7、1.8、1.9、2、2.1、2.2、2.3、2.4、2.5、2.6、2.7、2.8、2.9或者3重量%,所述活性组分以元素计。优选地,以加氢催化剂的总量为基准,所述活性组分的含量优选为0.5-3重量%。更优选地,以加氢催化剂的总量为基准,所述活性组分的含量优选为0.8-3重量%。
19.根据本发明的加氢催化剂,所述助剂组分为镍、铁和钴中的一种或两种以上。根据本发明的加氢催化剂,将钌与助剂组分组合使用,二者相互协同作用,能有效地促进催化剂活性的改善。根据本发明的加氢催化剂,以加氢催化剂的总量为基准,所述助剂组分的含量优选为0.3-3重量%,例如可以为:0.3、0.4、0.5、0.6、0.7、0.8、0.9、1、1.1、1.2、1.3、1.4、1.5、1.6、1.7、1.8、1.9、2、2.1、2.2、2.3、2.4、2.5、2.6、2.7、2.8、2.9或者3重量%,所述助剂组分以元素计。
20.根据本发明的加氢催化剂,在一种优选的实施方式中,所述助剂组分与所述活性组分的摩尔比为0.1-25∶1。根据该优选的实施方式能进一步提高加氢催化剂的催化活性。根据该优选的实施方式,在所述助剂组分为镍时,助剂组分与活性组分的摩尔比优选为0.5-1.5∶1,更优选为0.8-1.2∶1。根据该优选的实施方式,在所述助剂组分为钴时,助剂组分与活性组分的摩尔比优选为0.1-0.5∶1,更优选为0.12-0.25∶1。根据该优选的实施方式,在所述助剂组分为铁时,助剂组分与活性组分的摩尔比优选为10-25∶1,更优选为15-20∶1。
21.根据本发明的加氢催化剂,所述载体优选为活性炭、氧化硅、氧化钛和氧化锆中的一种或两种以上。在一种优选的实施方式中,所述助剂组分为镍,所述载体为活性炭和/或氧化钛。在另一种优选的实施方式中,所述助剂组分为铁,所述载体为氧化锆。在又一种优选的实施方式中,所述助剂组分为钴,所述载体为氧化硅。
22.根据本发明的加氢催化剂还含有碱金属元素,以加氢催化剂的总量为基准,所述碱金属元素的重量含量可以为10-1000ppm,优选为50-800ppm,更优选为80-600ppm,进一步优选为100-550ppm,所述碱金属金属元素以元素计。
23.本发明中,加氢催化剂中活性组分和助剂组分的含量采用x射线荧光光谱法测定,碱金属元素的含量采用电感耦合等离子体发射光谱法测定。
24.根据本发明的第二个方面,本发明提供了一种加氢催化剂的制备方法,该方法包括步骤(1):将载体与含有碱金属化合物的溶液接触,得到改性载体。根据本发明的方法,在将载体负载活性组分和助剂组分之前与含有碱金属化合物的溶液接触,在载体上引入碱金属,能明显提高加氢催化剂的催化活性。根据本发明的制备方法,最终制备的加氢催化剂中,所述碱金属元素的重量含量可以为10-1000ppm,优选为50-800ppm,更优选为80-600ppm,进一步优选为100-550ppm,所述碱金属元素以元素计。
25.步骤(1)中,所述碱金属化合物优选为碱金属氢氧化物,更优选为氢氧化钠、氢氧化钾和氢氧化锂中的一种或两种以上,进一步优选为氢氧化钠。
26.所述含有碱金属化合物的溶液的溶剂可以为水和/或c
1-c4的醇,优选为水。
27.步骤(1)中,载体与含有碱金属化合物的溶液接触的方法可以为常规方法,例如:浸渍、喷淋中的一种或两种以上的组合,优选为浸渍。所述浸渍可以为等体积浸渍,也可以为过量浸渍。所述浸渍的次数可以为一次,也可以为两次以上。在所述浸渍的次数为两次以上时,每次浸渍完成之后可以脱除载体上的挥发性组分。
28.载体与含有碱金属化合物的溶液可以在常规条件下进行接触。在一种优选的实施方式中,载体与含有碱金属化合物的溶液在20-60℃的温度下进行接触,例如:20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59或者60℃的温度下进行接触。所述接触的持续时间可以为2-20小时,优选为2-10小时。
29.步骤(1)中,载体与含有碱金属化合物的溶液接触后进行洗涤。可以将接触得到的固体物质用水进行洗涤。所述洗涤的条件优选为使得洗涤流出液(即,洗涤出水)的ph值为7.2-7.5。
30.将载体与含有碱金属氢氧化物的溶液接触得到的负载有所述溶液的载体可以采用常规方法脱除负载在载体上的挥发性组分,得到改性载体。具体地,可以将负载有所述溶液的载体进行干燥得到改性载体。所述干燥优选在低于150℃的温度下进行。在一种优选的
实施方式中,所述干燥在80-120℃的温度下进行。所述干燥的持续时间可以为4-20小时,优选为5-15小时。所述干燥可以在常压(即,1标准大气压)下进行,也可以在降低压力的条件下进行。
31.根据本发明的制备方法,包括步骤(2):将所述改性载体与含有活性组分前身物和助剂组分前身物的溶液接触,得到负载有活性组分前身物和助剂组分前身物的负载载体,脱除所述负载载体中的至少部分挥发性组分后进行焙烧,得到加氢催化剂前体。
32.步骤(2)中,所述活性组分为钌。本发明中,术语“活性组分前身物”是指在催化剂制备过程中能在催化剂中形成活性组分的物质。所述活性组分前身物优选为氯化钌、硝酸钌和乙酸钌中的一种或两种以上。所述助剂组分为镍、铁和钴中的一种或两种以上。本发明中,术语“助剂组分前身物”是指在催化剂制备过程中能在催化剂中形成助剂组分的物质。所述助剂组分前身物优选为助剂组分的硝酸盐、硫酸盐、甲酸盐、乙酸盐和氯化物中的一种或两种以上,其具体实例可以包括但不限于硝酸镍、硫酸镍、乙酸镍、氯化镍、硝酸钴、硫酸钴、乙酸钴、氯化钴、硝酸铁、硫酸铁、乙酸铁和氯化铁中的一种或两种以上。
33.步骤(2)中,含有活性组分前身物和助剂组分前身物的溶液的溶剂可以为水和/或c
1-c4的醇,优选为水。
34.含有活性组分前身物和助剂组分前身物的溶液中,活性组分前身物的含量优选为1
×
10-5
mol/ml至20
×
10-5
mol/ml,更优选为1.1
×
10-5
mol/ml至15
×
10-5
mol/ml,助剂组分前身物的含量优选为0.5
×
10-5
mol/ml至15
×
10-5
mol/ml,更优选为1
×
10-5
mol/ml至10
×
10-5
mol/ml,进一步优选为2
×
10-5
mol/ml至8
×
10-5
mol/ml。根据本发明的方法,步骤(1)中采用的碱金属化合物与步骤(2)中采用的活性组分前身物和助剂组分前身物的总量的摩尔比可以为1-8∶1,优选为1.5-6∶1,更优选为3-5∶1。
35.根据本发明的方法,所述活性组分前身物和助剂组分前身物以能在载体上引入满足要求的活性组分和助剂组分为准。根据本发明的方法,所述活性组分和所述助剂组分在载体上的负载量使得以最终制备的加氢催化剂的总量为基准,所述活性组分的含量优选为0.3-3重量%,更优选为0.5-3重量%,进一步优选为0.8-3重量%;所述助剂组分的含量优选为0.3-3重量%。根据本发明的方法,最终制备的加氢催化剂中,助剂组分与活性组分的摩尔比优选为0.1-25∶1。在所述助剂组分为镍时,最终制备的加氢催化剂中,助剂组分与活性组分的摩尔比优选为0.5-1.5∶1,更优选为0.8-1.2∶1。在所述助剂组分为钴时,最终制备的加氢催化剂中,助剂组分与活性组分的摩尔比优选为0.1-0.5∶1,更优选为0.12-0.25∶1。在所述助剂组分为铁时,最终制备的加氢催化剂中,助剂组分与活性组分的摩尔比优选为10-25∶1,更优选为15-20∶1。
36.步骤(2)中,载体与所述溶液接触的方法可以为常规方法,例如:浸渍、喷淋中的一种或两种以上的组合,优选为浸渍。所述浸渍可以为等体积浸渍,也可以为过量浸渍。所述浸渍的次数可以为一次,也可以为两次以上,以能在载体上引入足量的活性组分和助剂组分为准。在所述浸渍的次数为两次以上时,每次浸渍完成之后可以脱除载体上的挥发性组分。
37.步骤(2)中,载体与所述溶液可以在常规条件下进行接触。在一种优选的实施方式中,载体与所述溶液在40-80℃的温度下进行接触,例如:40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、
74、75、76、77、78、79或者80℃的温度下进行接触。所述接触的持续时间可以为5-30小时,优选为10-20小时,更优选为15-20小时。
38.步骤(2)中,将载体与所述溶液接触得到的负载有所述溶液的载体可以采用常规方法脱除负载在载体上的挥发性组分。具体地,可以将负载有所述溶液的载体进行干燥得到改性载体。所述干燥优选在低于150℃的温度下进行。在一种优选的实施方式中,所述干燥在80-120℃的温度下进行。所述干燥的持续时间可以为4-20小时,优选为8-20小时。所述干燥可以在常压(即,1标准大气压)下进行,也可以在降低压力的条件下进行。
39.步骤(2)中,所述焙烧在不高于300℃的温度下进行,如150-300℃的温度下进行。优选地,所述焙烧在不高于250℃的温度下进行。与在更高的温度下进行焙烧相比,在不高于250℃的温度下进行焙烧,能明显提高最终制备的加氢催化剂的催化活性。更优选地,所述焙烧在150-250℃的温度下进行,例如可以在150、151、152、153、154、155、156、157、158、159、160、161、162、163、164、165、166、167、168、169、170、171、172、173、174、175、176、177、178、179、180、181、182、183、184、185、186、187、188、189、190、191、192、193、194、195、196、197、198、199、200、201、202、203、204、205、206、207、208、209、210、211、212、213、214、215、216、217、218、219、220、221、222、223、224、225、226、227、228、229、230、231、232、233、234、235、236、237、238、239、240、241、242、243、244、245、246、247、248、249或者250℃的温度下进行。所述焙烧的持续时间可以为2-10小时。所述焙烧可以在含氧气氛中进行,也可以在还原性气氛中进行。
40.根据本发明的制备方法,包括步骤(3):在还原反应条件下,将所述加氢催化剂前体与还原剂接触,得到所述加氢催化剂。
41.所述还原剂可以为足以将加氢催化剂前体中的活性组分元素和助剂组分元素还原的物质。在一种优选的实施方式中,所述还原剂为水合肼、硼氢化钠和甲醛中的一种或两种以上。在一个优选实例中,所述助剂组分为镍和/或铁,所述还原剂优选为水合肼和/或甲醛。根据该优选实例,在还原剂为水合肼和甲醛时,水合肼(水合肼以肼n2h4计)与甲醛的摩尔比优选为1∶2-6,更优选为1∶3-5。在另一个实例中,所述助剂组分为钴,所述还原剂优选为硼氢化钠。
42.所述还原剂的用量可以根据加氢催化剂前体中活性组分和助剂组分的含量进行选择,以能将加氢催化剂前体中的活性组分和助剂组分还原为准。一般地,以摩尔计,步骤(3)中的还原剂:(步骤(2)中的活性组分+步骤(2)中的助剂组分)=3-6∶1(即,还原剂与步骤(2)中活性组分前身物和助剂组分前身物的总量的摩尔比为3-6∶1),所述活性组分前身物以活性组分计,所述助剂组分前身物以助剂组分计。
43.所述还原可以在20-80℃的温度下进行,例如在20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79或者80℃的温度下进行。所述还原的持续时间可以根据还原的温度进行选择,例如可以为1-10小时。在一个优选实例中,所述助剂组分为镍和/或钴,所述还原在50-80℃的温度下进行,所述还原的持续时间优选为1-5小时。在另一个优选实例中,所述助剂组分为铁,所述还原在20-40℃的温度下进行,所述还原的持续时间优选为6-10小时。
44.步骤(3)中,脱除经还原的催化剂前体中的挥发性组分得到根据本发明的加氢催
化剂。可以将经还原的催化剂前体进行干燥,从而脱除经还原的催化剂前体中的挥发性组分。所述干燥可以在40-150℃的温度下进行,优选在50-120℃的温度下进行,更优选在60-100℃的温度下进行,进一步优选在70-90℃的温度下进行。所述干燥的持续时间可以根据干燥的温度进行选择,一般可以为5-24小时,优选为6-20小时,更优选为8-10小时。所述干燥可以在含氧气氛中进行(如空气气氛中进行),也可以在非氧化性气氛中进行,所述非氧化性气氛例如氮气气氛和/或零族气体气氛(如氩气)。在含氧气氛中进行干燥时,所述干燥优选在不超过100℃的温度下进行,例如在40-80℃的温度下进行,优选在60-80℃的温度下进行。所述干燥可以在常压(即,1标准大气压)下进行,也可以在降低压力的条件下进行,没有特别限定。
45.根据本发明的第三个方面,本发明提供了一种由本发明第二个方面所述的方法制备的加氢催化剂。
46.根据本发明的第四个方面,本发明提供了根据本发明第一个方面或者第三个方面所述的加氢催化剂作为苯甲酸加氢反应的催化剂的应用。所述苯甲酸加氢制备得到的产物为环己基甲酸。
47.根据本发明的加氢催化剂具有提高的催化活性,能在温和的加氢反应条件下,获得提高的催化活性;并且,根据本发明的加氢催化剂适于在固定床反应器中使用,能够实现苯甲酸加氢反应的连续稳定进行,满足工业化生产规模的要求。
48.根据本发明的第五个方面,本发明提供了一种环己基甲酸的制备方法,该方法包括在加氢反应条件下,在加氢催化剂存在下,将苯甲酸与氢气接触,其中,所述加氢催化剂为本发明第一个方面或者第三个方面所述的加氢催化剂。所述加氢催化剂的组成及其制备方法在前文已经进行了详细的说明,此处不再详述。
49.根据本发明的环己基甲酸的制备方法,作为加氢反应原料的苯甲酸加氢反应原料液的形式提供,所述加氢反应原料液中,苯甲酸的含量可以为10-40重量%。所述加氢反应原料液的溶剂可以为环己甲酸、乙醇和乙酸乙酯中的一种或两种以上,优选为环己甲酸。
50.根据本发明的环己基甲酸的制备方法,加氢催化剂具有较高的催化活性,即便在较低的温度下进行加氢反应也能获得良好的加氢反应效果。根据本发明的环己基甲酸的制备方法,加氢反应(即,在加氢催化剂存在下,将苯甲酸与氢气接触)的温度可以在不超过120℃的温度下进行,优选在60-120℃的温度下进行,例如可以在60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、99、100、101、102、103、104、105、106、107、108、109、110、111、112、113、114、115、116、117、118、119或者120℃的温度下进行。
51.根据本发明的环己基甲酸的制备方法,加氢反应可以在压力为1-5mpa的条件下进行,所述压力为表压。
52.根据本发明的环己基甲酸的制备方法,氢气与苯甲酸的摩尔比可以为4-50∶1,例如:4∶1、5∶1、6∶1、7∶1、8∶1、9∶1、10∶1、11∶1、12∶1、13∶1、14∶1、15∶1、16∶1、17∶1、18∶1、19∶1、20∶1、21∶1、22∶1、23∶1、24∶1、25∶1、26∶1、27∶1、28∶1、29∶1、30∶1、31∶1、32∶1、33∶1、34∶1、35∶1、36∶1、37∶1、38∶1、39∶1、40∶1、41∶1、42∶1、43∶1、44∶1、45∶1、46∶1、47∶1、48∶1、49∶1或者50∶1。
53.根据本发明的环己基甲酸的制备方法,加氢反应优选为连续反应,例如可以在固
定床反应器中进行。在固定床反应器中进行加氢反应时,所述苯甲酸的重时空速优选为0.2-2h-1
,例如:0.2、0.3、0.4、0.5、0.6、0.7、0.8、0.9、1、1.1、1.2、1.3、1.4、1.5、1.6、1.7、1.8、1.9或者2h-1
。
54.根据本发明的环己基甲酸的制备方法,加氢催化剂在与苯甲酸和氢气接触,催化苯甲酸和氢气进行加氢反应之前,优选进行补充还原。所述补充还原可以在加氢反应器外进行,也可以在加氢反应器内进行。在采用固定床反应器进行加氢反应时,优选在反应器内进行补充还原,此时,可以先向加氢反应器内通入氢气,而不向加氢反应器内提供苯甲酸,使得氢气与加氢催化剂接触进行补充还原。
55.所述补充还原可以在100-180℃的温度下进行。所述补充还原可以在压力为0-5mpa的条件下进行,优选在2-5mpa的条件下进行,所述压力为表压。所述补充还原的持续时间可以为4-10小时。
56.与现有技术相比,根据本发明的环己基甲酸的制备方法采用的催化剂中贵金属的含量降低,因而能有效地降低催化剂成本;并且,根据本发明的环己基甲酸的制备方法采用的加氢催化剂具有良好的低温活性,即便在较低的温度下进行加氢反应也能获得较好的加氢反应效果。另外,根据本发明的环己基甲酸的制备方法,能够实现连续化稳定操作,简化工艺流程,提高生产效率,实现环己基甲酸的连续化生产,产品质量好且稳定。
57.以下结合实施例详细说明本发明,但并不因此限制本发明的范围。
58.以下实施例和对比例中,催化剂中ru和助剂组分的含量采用x射线荧光光谱法测定,碱金属的含量采用电感耦合等离子体发射光谱法测定。
59.以下实施例和对比例中,采用气相色谱法测定反应器输出的反应液的组成,并根据测定的组成数据采用以下公式计算原料转化率和产物选择性,
60.原料转化率=(加入的原料的摩尔量一剩余的原料的摩尔量)/加入的原料的摩尔量
×
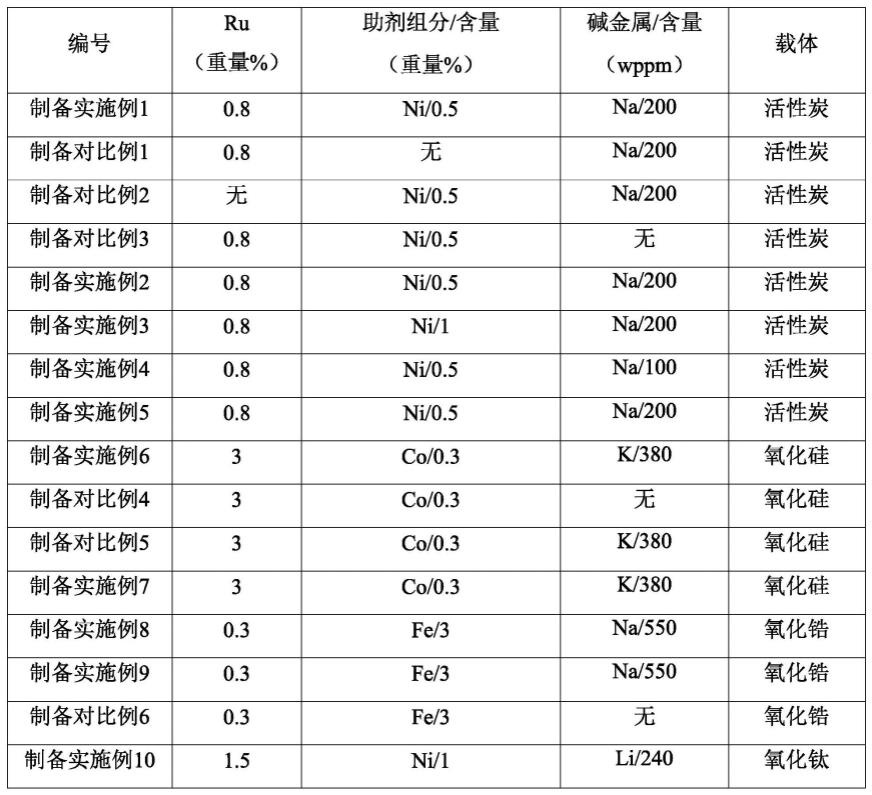
100%;
61.产物选择性=反应生成的产物的摩尔量/(加入的原料的摩尔量-剩余的原料的摩尔量)
×
100%。
62.以下实施例和对比例中,压力均为表压。
63.制备实施例1-10用于制备根据本发明的加氢催化剂。
64.制备实施例1
65.(1)在20℃的温度下,用25ml氢氧化钠水溶液浸渍活性炭(购自神华集团,比表面积为950m2/g)2小时,然后将经浸渍的活性炭用去离子水进行洗涤直至洗涤出水的ph值为7.2,接着将经洗涤的固体物质在100℃干燥10h,得到改性载体。
66.(2)在50℃的温度下,用25ml含rucl3和nicl2的水溶液浸渍步骤(1)制备的改性载体15小时,将经浸渍的改性载体在80℃干燥20h,然后在180℃空气气氛中焙烧10h,得到催化剂前体。其中,步骤(2)采用的水溶液中,rucl3的浓度为3.68
×
10-5
mol/ml,nicl2的浓度为3.45
×
10-5
mol/ml,步骤(1)中采用的naoh与步骤(2)中ru和ni总量的摩尔比为3∶1。
67.(3)将步骤(2)制备的催化剂前体置于水合肼水溶液(水合肼与ru和ni总量的摩尔比为4∶1,水合肼以肼计)中,在60℃的温度下反应4小时后,进行过滤,收集得到的固体物质用去离子水洗涤3次,然后在80℃的温度下于空气气氛中干燥8小时,从而得到根据本发明的加氢催化剂,具体组成见表1。
68.制备对比例1
69.采用与制备实施例1相同的方法制备加氢催化剂,不同的是,步骤(2)采用的水溶液不含nicl2。制备得到的加氢催化剂的组成在表1中列出。
70.制备对比例2
71.采用与制备实施例1相同的方法制备加氢催化剂,不同的是,步骤(2)采用的水溶液不含rucl3。制备得到的加氢催化剂的组成在表1中列出。
72.制备对比例3
73.采用与制备实施例1相同的方法制备加氢催化剂,不同的是,不进行步骤(1),直接将制备实施例1的步骤(1)采用的活性炭用于步骤(2)。制备得到的加氢催化剂的组成在表1中列出。
74.制备实施例2
75.采用与制备实施例1相同的方法制备加氢催化剂,不同的是,步骤(2)中,焙烧在300℃的温度下进行。制备得到的加氢催化剂的组成在表1中列出。
76.制备实施例3
77.采用与制备实施例1相同的方法制备加氢催化剂,不同的是,步骤(2)采用的水溶液中,nicl2的浓度为6.9
×
10-5
mol/ml。制备得到的加氢催化剂的组成在表1中列出。
78.制备实施例4
79.采用与制备实施例1相同的方法制备加氢催化剂,不同的是,步骤(1)中,改变氢氧化钠水溶液中氢氧化钠的浓度使得步骤(1)中采用的naoh与步骤(2)中ru和ni总量的摩尔比为1.5∶1。制备得到的加氢催化剂的组成在表1中列出。
80.制备实施例5
81.采用与制备实施例1相同的方法制备加氢催化剂,不同的是,步骤(3)中,采用等摩尔量硼氢化钠作为还原剂。制备得到的加氢催化剂的组成在表1中列出。
82.制备实施例6
83.(1)在60℃的温度下,用25ml氢氧化钾水溶液浸渍氧化硅(购自淄博恒齐粉体新材料有限公司,比表面积为180m2/g)2小时,然后用去离子水进行洗涤直至洗涤出水的ph值为7.4,接着将经洗涤的固体物质在120℃干燥5h,得到改性载体。
84.(2)在60℃的温度下,用25ml含硝酸钌、乙酸钌和乙酸钴的水溶液浸渍步骤(1)制备的改性载体15小时,将经浸渍的改性载体在110℃干燥10h,然后在250℃空气气氛中焙烧2h,得到催化剂前体。其中,步骤(2)采用的水溶液中,硝酸钌的浓度为5.94
×
10-5
mol/ml,乙酸钌的浓度为5.94
×
10-5
mol/ml,乙酸钴的浓度为2.04
×
10-6
mol/ml,步骤(1)中采用的koh与步骤(2)中ru和co总量的摩尔比为5∶1。
85.(3)将步骤(2)制备的催化剂前体置于硼氢化钠水溶液(硼氢化钠与ru和co总量的摩尔比为5∶1)中,在50℃的温度下反应5小时后,进行过滤,收集得到的固体物质用去离子水洗涤3次,然后在80℃的温度下于空气气氛中干燥8小时,从而得到根据本发明的加氢催化剂,具体组成见表1。
86.制备对比例4
87.采用与制备实施例6相同的方法制备加氢催化剂,不同的是,不进行步骤(1),直接将制备实施例1的步骤(1)采用的氧化硅用于步骤(2)。制备得到的加氢催化剂的组成在表1
中列出。
88.制备对比例5
89.采用与制备实施例6相同的方法制备加氢催化剂,不同的是,步骤(2)中,焙烧的温度为300℃。制备得到的加氢催化剂的组成在表1中列出。
90.制备实施例7
91.采用与制备实施例6相同的方法制备加氢催化剂,不同的是,步骤(3)中,硼氢化钠用等摩尔量的甲醛代替。制备得到的加氢催化剂的组成在表1中列出。
92.制备实施例8
93.(1)在50℃的温度下,用50ml氢氧化钠水溶液浸渍氧化锆(购自淄博启明星新材料股份有限公司,比表面积为120m2/g)4小时,然后用去离子水进行洗涤直至洗涤出水的ph值为7.5,接着将经洗涤的固体物质在80℃干燥14h,得到改性载体。
94.(2)在40℃的温度下,用25ml含硝酸钌和硝酸铁的水溶液浸渍步骤(1)制备的改性载体20小时,将经浸渍的改性载体在120℃干燥8h,然后在150℃空气气氛中焙烧6h,得到催化剂前体。其中,步骤(2)采用的水溶液中,硝酸钌的浓度为1.19
×
10-5
mol/ml,硝酸铁的浓度为2.15
×
10-4
mol/ml,步骤(1)中采用的naoh与步骤(2)中ru和fe总量的摩尔比为5∶1。
95.(3)将步骤(2)制备的催化剂前体置于含有水合肼和甲醛的水溶液(水合肼和甲醛的总量与ru和fe总量的摩尔比为6∶1,水合肼和甲醛的摩尔比为1∶4,水合肼以肼计)中,在20℃的温度下反应10小时后,进行过滤,收集得到的固体物质用去离子水洗涤3次,然后在70℃的温度下于空气气氛中干燥10小时,从而得到根据本发明的加氢催化剂,具体组成见表1。
96.制备实施例9
97.采用与制备实施例8相同的方法制备加氢催化剂,不同的是,步骤(3)中,还原剂水溶液不含水合肼(即,水合肼用等摩尔量的甲醛代替,还原剂的总摩尔量与制备实施例8相同)。制备得到的加氢催化剂的组成在表1中列出。
98.制备对比例6
99.采用与制备实施例8相同的方法制备加氢催化剂,不同的是,不进行步骤(1),直接将制备实施例1的步骤(1)采用的氧化锆用于步骤(2)。制备得到的加氢催化剂的组成在表1中列出。
100.制备实施例10
101.(1)在40℃的温度下,用50ml氢氧化锂水溶液浸渍氧化钛(购自淄博恒齐粉体新材料有限公司,比表面积为120m2/g)8小时,然后用去离子水进行洗涤直至洗涤出水的ph值为7.3,接着将经洗涤的固体物质在110℃干燥6h,得到改性载体。
102.(2)在80℃的温度下,用25ml含硝酸钌和硝酸镍的水溶液浸渍步骤(1)制备的改性载体20小时,将经浸渍的改性载体在100℃干燥12h,然后在200℃空气气氛中焙烧5h,得到催化剂前体。其中,步骤(2)采用的水溶液中,硝酸钌的浓度为5.94
×
10-5
mol/ml,硝酸镍的浓度为6.89
×
10-5
mol/ml,步骤(1)中采用的lioh与步骤(2)中ru和ni总量的摩尔比为4∶1。
103.(3)将步骤(2)制备的催化剂前体置于甲醛水溶液(甲醛与ru和ni总量的摩尔比为3∶1)中,在80℃的温度下反应1小时后,进行过滤,收集得到的固体物质用去离子水洗涤3次,然后在70℃的温度下于空气气氛中干燥10小时,从而得到根据本发明的加氢催化剂,具
体组成见表1。
104.表1
[0105][0106]
实施例1-16用于说明根据本发明的加氢催化剂的应用和环己基甲酸的制备方法。
[0107]
实施例1-12
[0108]
(1)加氢催化剂的装填
[0109]
首先在管式固定床加氢反应器底部装填底部惰性瓷球层起支撑作用,然后以散堆方式在瓷球上方装填加氢催化剂,加氢催化剂的装填高度与管式固定床加氢反应器中反应管的管径之比为10∶1,最后在床层上方再装填顶部惰性瓷球层,并安装反应器顶部封头。
[0110]
(2)补充还原
[0111]
向管式固定床加氢反应器中,通入氢气,在表2列出的条件下进行补充加氢反应。
[0112]
(3)加氢反应
[0113]
将苯甲酸溶液(溶剂为环己甲酸)通入管式固定床加氢反应器中,在表3列出的条件下连续进行72小时加氢反应,测定管式固定床加氢反应器输出的反应产物的组成,计算苯甲酸转化率和环己基甲酸选择性,结果在表3中列出。
[0114]
对比例1-3
[0115]
采用与实施例1相同的方法制备环己基甲酸,不同的是,分别采用制备对比例1-3制备的加氢催化剂,实验结果在表3中列出。
[0116]
对比例4-5
[0117]
采用与实施例6相同的方法制备环己基甲酸,不同的是,分别采用制备对比例4-5制备的加氢催化剂,实验结果在表3中列出。
[0118]
对比例6
[0119]
采用与实施例9相同的方法制备环己基甲酸,不同的是,采用制备对比例6制备的加氢催化剂,实验结果在表3中列出。
[0120]
表2
[0121]
[0122][0123]
实施例13-16
[0124]
采用与实施例1-12相同的方法装填催化剂并进行补充还原(其中,实施例13采用
与实施例1相同的方法进行补充还原,实施例14采用与实施例6相同的方法进行补充还原,实施例15采用与实施例8相同的方法进行补充还原,实施例16采用与实施例10相同的方法进行补充还原),在表4列出的条件下进行长时间连续加氢反应,反应过程连续监测从加氢反应器中流出的反应产物的组成并计算原料转化率和产物选择性,反应0.5小时和500小时的实验结果在表4中列出。
[0125]
实施例1-12的实验结果证实,根据本发明的加氢催化剂具有良好的低温活性,即便在较低的温度下进行加氢反应也能获得较好的加氢反应效果。
[0126]
实施例13-16的实验结果证实,根据本发明的加氢催化剂具有较长的使用寿命,因而根据本发明的环己基甲酸的制备方法能够实现连续化稳定操作,简化工艺流程,提高生产效率,实现环己基甲酸的连续化生产,产品质量好且稳定。
[0127]
以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。
[0128]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1