用于合成烷基硫醇的催化剂及其制备方法与流程
用于合成烷基硫醇的催化剂及其制备方法
1.本发明涉及一种用于制备烷基硫醇的tio2和/或zro2系催化剂及其制备方法。本发明还涉及一种在根据本发明的负载型催化剂存在下或在通过根据本发明的其制备方法获得的催化剂存在下,使烷基醇与硫化氢反应来制备烷基硫醇的方法。
2.烷基硫醇是用于制备具有高经济价值的产品的工业上重要的中间体。特别地,甲硫醇(ch3sh)是工业上重要的中间体,例如在甲硫氨酸、二甲基亚砜、二甲基砜或甲磺酸的合成中。过去开发了多种不同的合成路线,例如,硫化羰(cos)或二硫化碳(cs2)的氢化,在存在和不存在h2下用cs2对甲醇进行硫醇化,以及在基于氧化铝的催化剂的存在下用硫化氢(h2s)作为硫醇化剂对甲醇进行硫醇化。后一种方法代表了本领域方法的当前工业状态,其通常在气相中在300至500℃的温度和1至25巴的压力下进行。
3.由此获得的反应混合物含有所需的产物甲硫醇以及未反应的起始原料和副产物,例如二甲硫醚和二甲醚,以及对反应呈惰性的气体,例如甲烷、一氧化碳、氢气和氮。因此,必须从该反应混合物中分离出所形成的甲硫醇。
4.从气态反应混合物中分离甲硫醇通常是通过甲硫醇的缩合来实施的。在这里,冷却反应混合物的能量消耗是一个很大的成本因素。因此,为了该方法的良好经济性,必须具有高转化率和高选择性来形成甲硫醇,以保持尽可能低的能量输入和投资成本。
5.通过增加硫化氢与甲醇的摩尔比可获得活性和选择性的改进。通常使用介于1和10之间的摩尔比。然而,高摩尔比也意味着反应混合物中硫化氢高度过量,因此需要循环大量气体。为了减少为此所需的能量输入,硫化氢与甲醇的比率因此应仅略微偏离1。
6.为了提高活性和选择性,设计了特定的al2o3系催化剂。一般认为,氧化铝,特别是γ-al2o3原则上是用于甲醇的硫醇化以得到甲硫醇的催化剂。然而,氧化铝的活性太高,因此催化反应不会在所需的产物甲硫醇处停止。更确切地,非常活泼的氧化铝还催化甲硫醇进一步反应生成二甲硫醚。因此,通常将氧化铝与碱金属钨酸盐如钨酸钾或钨酸铯混合以降低其活性,这会导致形成甲硫醇的选择性增加,且形成副产物诸如二甲硫醚的选择性降低。在由此获得的催化剂中,氧化铝也称为载体或催化剂载体,而碱金属钨酸盐则称为助催化剂。基于催化剂的总重量,钨酸盐的比例通常高达约20重量%,例如在美国专利第2,820,062号中描述的。该文献中公开的催化剂在烷硫醇的生产中在400℃的反应温度和2∶1的硫化氢与烷基醇的摩尔比下,提供了良好的活性和选择性。
7.在涉及用碱金属钨酸盐溶液多次浸渍载体材料的相对复杂的制备过程中,催化剂中碱金属钨酸盐的比例可以增加至25重量%或更高。美国专利第5,852,219号公开了使用钨酸铯(cs2wo4)代替钨酸钾(k2wo4)作为助催化剂的优点。因此,可以在良好的选择性的情况下同时获得增加的活性。根据mashkina等人(react.kinet.catal.lett.,第36卷第1期,159-164(1988)),由烷基醇和硫化氢形成烷基硫醇的最佳选择性是用碱/钨比等于2∶1的催化剂实现的。用含有化学计量比为2∶1的铯和钨的溶液浸渍氧化铝载体允许实现基于催化剂的总重量高达40重量%的助催化剂负载,如美国专利第5,852,219号中所描述的。将碱金属钨酸盐的浓度增加至25重量%或更高导致形成甲硫醇的选择性增加。然而,缺点是同时催化剂的活性降低。
8.如美国专利申请2009/306432 a1中所述,用包含非化学计量比的小于2∶1的铯和钨的溶液对载体材料进行多次浸渍允许将催化剂与助催化剂的负载增加至基于催化剂总重量25重量%或更高的值。虽然通过在浸渍溶液中使用非化学计量比的铯和钨可以将铝与助催化剂的负载增加至大于35重量%以上,但在如此高负载的情况下,活性或选择性不再有显著增加。特别地,基于催化剂的总重量,活性和选择性甚至会随着负载高于45重量%而降低。另一缺点是通过多次浸渍方法生产的催化剂在整个催化剂主体中没有均匀的铯和钨分布。然而,为了在催化反应中实现高活性和高选择性,认为必须使催化活性组分在整个催化剂主体中均匀分布。
9.美国专利申请2014/357897 a1公开了在整个催化剂成型体中碱金属和钨分布均匀的催化剂。这些催化剂是通过将载体材料与氧化性钨材料如钨酸和至少一种单独的碱金属化合物如氢氧化铯混合,以得到催化剂物质,随后将所述催化剂物质成型来生产的。基于催化剂的总重量,由此获得的催化剂具有45重量%或更高的助催化剂负载。这允许进一步提高形成甲硫醇的收率和选择性。然而,所述催化剂的高收率和选择性不能持久,因此不适合用于工业过程。
10.wo 2017/114858 a1公开了另一种制备用于制备烷硫醇的催化剂的方法。该制备方法代表了普通浸渍程序和美国专利申请2014/357897 a1的混合或成型方法的组合。由此获得的催化剂的活性和甲硫醇选择性与其他催化剂相当。然而,相当复杂的制备方法并没有带来性能的提升。
11.如上所述,为改进用于甲醇硫醇化的催化剂的活性和选择性做出了相当大的努力。然而,催化剂主体系似乎不允许甲醇硫醇化的任何进一步改进。因此,有待解决的问题是提供一种用于甲醇硫醇化的改进的催化剂,其与现有技术的催化剂相比具有改进的选择性。
12.令人惊奇地发现,通过使用不同于现有技术的载体的催化剂载体解决了该问题。这种特殊的催化剂载体负载有碱金属氧化物。
13.因此,本发明的一个目的是一种催化剂,其包含载体和基于催化剂的总重量5至20重量%的助催化剂,或由它们组成,其中所述载体包含二氧化钛、二氧化锆和/或其混合物,或者由它们组成,并且助催化剂是碱金属氧化物。
14.基于催化剂的总重量计,助催化剂的5-20重量%的含量偏离仍然在本发明的范围内,只要它们仍然导致本发明的效果。
15.根据本发明,催化剂的载体包含二氧化钛、二氧化锆和/或它们的混合物,或由它们组成。因此,载体不一定必须由二氧化钛、二氧化锆和/或它们的混合物组成。这考虑了材料仍包含粘合剂、填料或任何其他来自其生产的成分的情况。然而,独立于此的主要量的材料应该是二氧化钛、二氧化锆和/或它们的混合物。优选地,催化剂的载体包含至少50重量%,特别是55-100重量%、60-100重量%、65-100重量%、70-100重量%、75-100重量%、80-100重量%、85-100重量%、90-100重量%或95-100重量%的二氧化钛、二氧化锆和/或它们的混合物。在极端情况下,载体由二氧化钛、二氧化锆和/或它们的混合物组成。
16.根据本发明的载体材料不仅在其元素组成上而且在结构上都不同于现有技术的载体。它们包含四方相或由四方相组成。相比之下,现有技术中作为催化剂载体的纯γ-al2o3具有立方相。
17.在一个实施方案中,根据本发明的催化剂的至少一部分载体具有四方相。优选地,根据本发明的催化剂的载体由四方相组成。
18.现有技术的催化剂通常含有碱金属钨酸盐作为助催化剂。然而,发现不一定需要碱金属钨酸盐来提供对在烷基醇硫醇化反应中形成烷基硫醇具有所需选择性的催化剂。更确切地,使用碱金属氧化物作为助催化剂已经足够。在催化剂的启动阶段,碱金属氧化物被硫化。
19.原则上,本发明对于助催化剂的特定碱金属的选择没有限制。因此碱金属可以是任何已知的碱金属,优选钠、钾、铯或铷。然而,铯是最提高催化剂选择性的碱金属。
20.在根据本发明的催化剂的进一步的实施方案中,碱金属因此是钠、钾、铯或铷。
21.在根据本发明的催化剂的另一个实施方案中,助催化剂是氧化铯。
22.与现有技术的催化剂相比,根据本发明的负载cs的催化剂提供甲硫醇选择性的显著提高。对于具有10重量%cs的负载的zro2系催化剂,甲硫醇的选择性增加到s
ch3sh,300℃
=99.9%至s
ch3sh,360℃
=99.1%的范围。发现的唯一副产物是选择性为s
dms,300℃
=0.1%至s
dms,360℃
=0.9%的二甲硫醚。将cs负载增加至20重量%导致介于s
ch3sh,300℃
=99.9%和s
ch3sh,360℃
=99.4%之间的甚至更高的选择性。对于tio2系催化剂发现了类似的结果:在10重量%cs的情况下,对甲硫醇的选择性增加至介于s
ch3sh,300℃
=99.9%至s
ch3sh,360℃
=99.4%之间的范围。再次,发现的唯一副产物是选择性为s
dms,300℃
=0.1%至s
dms,360℃
=0.6%的二甲硫醚。将cs负载增加至20重量%再次导致介于s
ch3sh,300℃
=99.9%和s
ch3sh,360℃
=99.5%之间的甚至更高的选择性。根据本发明的催化剂所实现的甲硫醇选择性高于现有技术催化剂所实现的甲硫醇选择性的绝对值2%以上。相比之下,wo 2013/092129 a1的最佳催化剂最多提供97.9%的甲硫醇选择性。
23.基于催化剂的总重量,根据本发明的催化剂包含5至20重量%的助催化剂。在催化剂是壳催化剂的情况下,5至20重量%的量与壳的组成有关。
24.原则上,根据本发明的催化剂在其形状方面不受限制。以其最简单的形式,所述催化剂是负载型催化剂,其中氧化和/或硫化物形式的碱金属已被施加到包含二氧化锆和/或二氧化钛的载体上。在那种情况下,将包含具有氧化和/或硫化物形式的碱金属的化合物的浸渍水溶液直接浸渍到载体上,以制备负载型催化剂形式的催化剂。载体在具体尺寸方面不受限制。例如,它可以以粒径小于1000μm、小于500μm,至多250μm如125至250μm,或至多125μm如25至125μm的粉末形式存在,所述粒径根据国际标准iso 13320(2009)通过激光散射在湿分散体中测定。因此,根据本发明的催化剂也可以以挤出物或粒料的形式存在,其通过用含有包含氧化和/或硫化物形式的碱金属的化合物的水溶液浸渍粉末载体,然后干燥和煅烧来获得,其中煅烧将氧化和/或硫化物形式的碱金属转化为碱金属氧化物。将由此获得的催化物质与粘合剂混合并进行成型,以提供全催化剂。通常,由此获得的催化剂再次经受煅烧(其中烧掉任何粘合剂),任选地随后在100至200℃的温度下进行回火步骤。
25.在进一步的实施方案中,根据本发明的催化剂是全催化剂。
26.当生产核-壳催化剂时,用含有包含氧化和/或硫化物形式的碱金属的化合物的水溶液浸渍上述粉末载体。任选地将由此获得的混合物煅烧,与粘合剂混合并以球体的形式施加到惰性载体核上(例如由陶瓷制成),然后进行煅烧并任选回火以得到核-壳催化剂。
27.在另一个实施方案中,根据本发明的催化剂是核-壳催化剂。
28.根据本发明的催化剂仅需要在载体上存在氧化和/或硫化物形式的碱金属,而不一定要存在含有氧化形式的钨的化合物,例如钨酸或钨酸盐。与现有技术的方法相比,这也显著简化了催化剂的制备,其中必须满足碱与钨的特定比例。
29.本发明的另一个目的是制备根据本发明的催化剂的方法,其包括以下步骤:
30.a)用包含可溶性碱化合物(alkali compound)的水溶液浸渍包含二氧化钛、二氧化锆和/或它们的混合物或由其组成的载体以提供浸渍的载体,
31.b)干燥从步骤a)获得的浸渍载体,和
32.c)煅烧从步骤b)获得的干燥的浸渍载体以提供催化剂。
33.为了将浸渍溶液施加到载体上,可以使用各种浸渍技术,诸如浸入浸渍、喷雾浸渍、真空浸渍和孔隙体积浸渍。这也使得浸渍可以进行多于一次。在成型件的情况下,所选择的浸渍方法必须能够在成型件的整个横截面上以良好的均匀性施加所需负载量的助催化剂。浸渍溶液优选通过喷雾或真空浸渍以一或两步施加到成型件上。在喷雾浸渍中,水性浸渍溶液被喷洒到载体上。在真空浸渍中,使用真空泵在装有成型件的容器中产生减压。通过打开与水性浸渍溶液的连接,将溶液吸入所述容器中,直到成型件的整体装料(charge)被所述溶液覆盖。在0.2至2小时的浸渍期后,将未被材料吸收的溶液排出或倒出。通过在室温下预干燥1至10小时,成型件的横截面上的初始浓度梯度可以在很大程度上均衡。因此,改进了催化剂颗粒横截面上的浸渍均匀性。将由此获得的催化剂前体优选干燥过夜,例如在50至100℃,优选在60至80℃下干燥1至10小时,以去除残留水分。然后在300至600℃,优选在420至480℃下进行1至20小时,优选1至5小时的煅烧。结果,来自浸渍溶液的氧化和/或硫化物形式的碱金属被转移到碱金属氧化物中作为助催化剂,所述助催化剂固定在载体上,并且来自浸渍溶液的阴离子被破坏和驱除。在干燥和煅烧期间,可以任选地使气流流过用于催化剂前体的载体的装料,这改进了残留水分和分解气体的去除。与煅烧的时段和温度的明确提及的值的偏差在本发明的范围内,只要它们导致与明确提及的值相同的效果品质。
34.根据本发明的方法在浸渍溶液中碱金属化合物的选择方面不受限制。唯一的要求是碱金属化合物必须在水中具有足够的溶解度,以便使载体负载有所需浓度的碱金属,并且阴离子在煅烧步骤期间容易分解。因此优选使用碱金属氢氧化物、碱金属乙酸盐、碱金属碳酸盐或碱金属硝酸盐。
35.尽管如此,也可以使用在水中具有相对低溶解度的碱金属化合物。当碱金属化合物在水中的低溶解度不允许在单个浸渍步骤中获得所需的碱金属负载时,载体的浸渍也可以在多个步骤中进行,特别是在两个步骤中。例如,在第一步骤中使用的浸渍溶液于是含有包含氧化和/或硫化物形式的碱金属化合物的总量的三分之一至三分之二,其余的量在第二步骤或任何进一步的步骤中被施加到载体上。在多步骤例如在两步骤程序中,第一步骤中获得的中间产物任选地不煅烧。除此之外,第二步骤中进行与针对一步骤法所述相同的浸渍、干燥和煅烧程序。
36.在制备根据本发明的催化剂的方法的一个实施方案中,步骤a)至b)或a)至c)重复至少一次。
37.由此获得的催化剂,特别是从步骤c)获得的催化剂可以与粘合剂混合,然后经受成型过程,诸如挤出或造粒,以得到全催化剂。对由此获得的挤出物或粒料进行最终煅烧和
任选存在的回火。
38.在另一个实施方案中,用于制备催化剂的方法进一步包括步骤:
39.d1)使从制备催化剂的方法的步骤c)中获得的催化剂成型以得到全催化剂。
40.或者,也可以将来自步骤c)的催化剂施加到核上以得到核-壳催化剂。为此,将从步骤c)获得的催化剂悬浮在溶剂中,优选水中,与粘合剂混合,并且将由此获得的混合物施加到惰性核(例如,由陶瓷材料制成)上,例如通过喷雾干燥,随后煅烧以除去溶剂并烧掉粘合剂,并任选地进行回火。
41.在替代的实施方案中,用于制备催化剂的方法进一步包括以下步骤:
42.d2)将从制备催化剂的方法的步骤c)中获得的催化剂施加到核上以提供核-壳催化剂。
43.根据本发明的催化剂以及通过根据本发明的方法获得的催化剂适用于烷基醇与硫化氢的催化反应以得到烷基硫醇,这也称为烷基醇硫醇化。
44.因此,本发明的进一步的目的是一种制备烷基硫醇的方法,其中在根据本发明的催化剂或通过根据本发明的方法获得的催化剂的存在下使烷基醇与硫化氢反应。
45.原则上,根据本发明的硫醇化方法不限于使用特定的烷基醇或制备特定的烷基硫醇。然而,经济上最相关的烷基醇是甲硫醇。
46.在根据本发明的硫醇化方法的实施方案中,待反应的烷基醇因此是甲醇,并且待制备的烷基硫醇是甲硫醇。
47.通过以下项目进一步说明本发明:
48.1.一种包含载体和助催化剂的催化剂,其中载体包含二氧化钛、二氧化锆和/或它们的混合物,并且助催化剂是氧化和/或硫化物形式的碱金属。
49.2.根据项目1的催化剂,其中至少一部分载体具有四方相。
50.3.根据项目1或2的催化剂,其中助催化剂是碱金属氧化物和/或碱金属硫化物。
51.4.根据项目1至3中任一项的催化剂,其中碱金属是钠、钾、铯或铷。
52.5.根据项目1至4中任一项的催化剂,其中基于催化剂的总重量,催化剂包含至多25重量%的助催化剂。
53.6.根据项目1至5中任一项的催化剂,其中基于催化剂的总重量,催化剂包含5至20重量%的助催化剂。
54.7.根据项目1至6中任一项的催化剂,其中助催化剂是氧化铯和/或硫化铯,并且基于催化剂的总重量,催化剂包含5至20重量%的所述助催化剂。
55.8.根据项目1至7中任一项的催化剂,其中催化剂是全催化剂。
56.9.根据项目1至8中任一项的催化剂,其中催化剂是核-壳催化剂。
57.10.一种制备根据项目1至7中任一项的负载型催化剂的方法,包括以下步骤:
58.a)用包含可溶性碱化合物的水溶液浸渍包含二氧化钛、二氧化锆和/或它们的混合物的载体,
59.b)干燥从步骤a)获得的浸渍载体,和
60.c)煅烧步骤b)的干燥的浸渍载体以提供催化剂。
61.11.根据项目10的方法,其中步骤a)至c)重复至少一次。
62.12.根据项目10或11的方法,进一步包括以下步骤:
63.d1)使从步骤c)获得的催化剂成型以得到全催化剂。
64.13.根据项目10或11的方法,进一步包括以下步骤:
65.d2)将从步骤c)中获得的催化剂施加到核上以提供核-壳催化剂。
66.14.一种制备烷基硫醇的方法,其中在根据项目1至9中任一项的催化剂或通过根据项目10至13中任一项的方法获得的催化剂的存在下使烷基醇与硫化氢反应。
67.15.根据项目14的方法,其中待反应的烷基醇是甲醇,并且待制备的烷基硫醇是甲硫醇。
附图说明
68.图1所示为纯金属氧化物(a)和负载有5重量%cs(b)、10重量%cs(c)、15重量%cs(d)和20重量%cs(e)的金属氧化物的xrd图案,其中γ表示纯γ-al2o3的特征信号,t表示四方zro2,a表示锐钛矿(tio2),并且cs表示cs2co3。
69.图2所示为50℃下纯金属氧化物上所吸收的吡啶的差减红外光谱,其中实线代表0.1毫巴的吡啶分压时所取的红外光谱,断线(broken line)代表在10-7
毫巴下抽真空后所取的红外光谱。
70.图3所示为γ-al2o3、zro2和tio2的oh振动区域的示差光谱,其中实线代表0.1毫巴的吡啶分压时所取的红外光谱,断线代表在10-7
毫巴下抽真空后所取的红外光谱。
71.图4所示为cs负载为10或20重量%的金属氧化物在50℃下的差减红外光谱,其中实线代表0.1毫巴的吡啶分压时所取的红外光谱,断线代表在10-7
毫巴下抽真空后所取的红外光谱。
72.图5所示为在5毫巴的co分压和在-150℃下,γ-al2o3(左)、zro2(中)和tio2(右)上co吸附的红外光谱,其中(a)纯金属氧化物,(b)cs负载为10重量%,(c)cs负载为20重量%。
73.图6所示为在0.1毫巴的甲醇分压和在50℃下,al2o3(左)、zro2(中)和tio2(左)上所吸附甲醇的红外光谱,其中(a)纯金属氧化物,(b)cs负载为10重量%,(c)cs负载为20重量%。
74.图7所示为在甲醇分压和温度为(a)50℃和0.1毫巴,(b)50℃和1毫巴,(c)100℃和1毫巴,(d)150℃和1毫巴,(e)200℃和1巴,(f)250℃和1毫巴,和(g)300℃和1毫巴下,纯金属氧化物γ-al2o3(左)、zro2(中)和tio2(右)上甲醇吸收的红外光谱。
75.图8所示为在甲醇分压和温度为(a)50℃和0.1毫巴,(b)50℃和1毫巴,(c)100℃和1毫巴,(d)150℃和1毫巴,(e)200℃和1毫巴,(f)250℃和1毫巴,和(g)300℃和1毫巴下,含有10重量%和20重量%cs的γ-al2o3(左)、含有10重量%和20重量%cs的zro2(中)和含有10重量%和20重量%cs的tio2(右)上的甲醇的红外光谱。
76.图9所示为对于纯金属氧化物(带立方形的实线)、cs负载为10重量%(带圆圈的虚线)和cs负载为20重量%(带三角形的断线),在300和360℃之间的温度下在γ-al2o3(左)、zro2(中)和tio2(右)上甲硫醇形成的初始速率。
77.图10所示为在360℃下在纯金属氧化物γ-al2o3(左)、zro2(中)和tio2(右)上作为甲醇转化率函数的甲硫醇(立方形)、二甲醚(圆圈)和二甲硫醚(三角形)的收率。
78.图11所示为在300和360℃之间的温度下在含有10重量%和20重量%cs的γ-al2o3(左)、含有10重量%和20重量%cs的zro2(中)和含有10重量%和20重量%cs的tio2(右)上
的作为甲醇转化率函数的甲硫醇(立方形)、二甲醚(圆圈)和二甲硫醚(三角形)的收率。
79.图12所示为甲硫醇形成速率对甲醇(立方形;y=0.3x+1.7)和硫化氢(三角形;y=0.5x+1.9)中的γ-al2o3、甲醇(立方形;y=0.2x
–
0.3)和硫化氢(三角形;y=0.4x
–
0.6)中的zro2,和甲醇(立方形;y=0.3x+1.2)和硫化氢(三角形;y=0.5x+1.7)中的tio2的依赖性,浓度以mol/l为单位。
80.图13所示为甲硫醇形成速率对具有不同铯负载的催化剂的依赖性:10重量%的cs(第一排),在由立方形表示的甲醇中(γ-al2o3:y=0.4x+1.0;zro2:y=0.5x+1.5;且tio2:y=0.6x+2.3),和在由三角形表示的硫化氢中(γ-al2o3:y=0.4x+0.6;zro2:y=0.3x-0.01;且tio2:y=0.2x+0.2)。20重量%的cs(第二排),在由立方形表示的甲醇中(γ-al2o3:y=0.3x+1.0;zro2:0.6x+2.3;且tio2:y=0.5x+2.0),和在由三角形表示的硫化氢中(γ-al2o3:y=0.5x+1.3;zro2:y=0.3x+0.5;且tio2:y=0.2x+0.3),浓度以mol/l表示。
81.图14所示为二甲醚形成速率对甲醇中(由立方形表示;y=1.5x+11.0)和硫化氢中(由三角形表示,y=0.0x)的纯γ-al2o3的依赖性,对甲醇中(以立方体表示;y=0.7x)的纯zro2的依赖性和对纯tio2(由立方形表示;y=0.7x+2.3)的依赖性,浓度以mol/l表示。
82.图15所示为在300、320、340和360℃(实心方块)的温度下,在负载有csws2(钨含量为5.1重量%,铯含量为20.6重量%)的γ-al2o3上的甲硫醇形成的初始速率。
实施例:
83.1.根据本发明的cs负载的金属氧化物的制备
84.通过用滴加到搅拌的固体中的乙酸铯水溶液,对各自具有0.125-0.25mm的晶粒大小的市售金属氧化物γ-al2o3(spheralite 101,axens)、tio2(hombikat 100uv,sachtleben)和zro2(sz 61152,norpro)进行初始润湿浸渍来制备cs负载为5、10、15和20重量%(基于催化剂的总重量)的催化剂。对于每种cs负载,制备含有所需量乙酸铯的不同浸渍溶液,以提供所需的cs负载。将76mg的乙酸铯(sigma aldrich,≥99.99%)溶解在0.5ml h2o/1g载体中以得到5重量%的cs负载,分别地对于10重量%的cs负载为160.5mg的乙酸铯,对于15重量%的cs负载为255mg的乙酸铯,并且对于20重量%的cs负载为361.0mg的乙酸铯。将浸渍的金属氧化物在70℃下干燥过夜,随后在以100ml/min流速的流动合成空气中并以0.5℃/min的升温斜率(ramp)实施在400℃下煅烧2小时。在其用于催化测试之前,所有样品都通过在360℃下在以20ml/min流速的h2s中处理2小时来活化。
85.2.所制备催化剂的表征
86.2.1元素组成和表面积测定
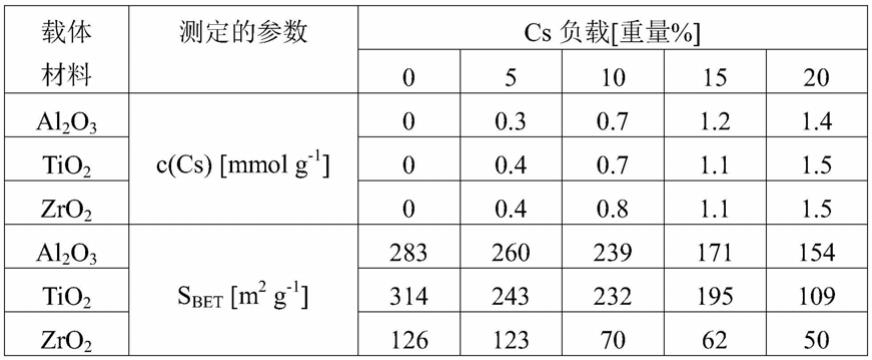
87.通过原子吸收光谱(aas)确定根据本发明制备的催化剂的元素组成。在unicam 939aa-光谱仪上进行测量。为了确定结构特性,在美国pmi公司(porous materials inc.)的bet-121比表面积分析仪上进行n2物理吸附。在真空下在250℃下活化2小时后,在77.4k的温度下吸附n2。使用bet方法计算表面积。所有制备的催化剂的元素分析和表面测定的结果总结在下表1中。
[0088][0089]
表1:所有制备的催化剂的元素分析和表面测定的结果
[0090]
结果表明,三种类型的载体材料中的每一种都实现了可比较的cs负载。一般而言,所制备的负载型催化剂的比表面积随着cs负载的增加而降低。这可归因于催化剂密度的增加和cs对表面的覆盖,两者均导致表面积损失。
[0091]
下表2总结了用于制备催化剂的不同浸渍溶液中的乙酸铯的不同质量、这些浸渍溶液中cs
+
的质量(忽略乙酸根抗衡离子的重量)、催化剂的质量(载体+cs
+
)、所制备的催化剂中cs
+
的理论浓度(c
th
(m(铯)/m(催化剂))、利用元素分析发现的所制备的催化剂中cs
+
的浓度(c
ea
(m(铯)/m(催化剂))。
[0092][0093]
表2:对所制备的催化剂及其铯浓度的概述。
[0094]
2.2晶体结构
[0095]
所有载体和所有催化剂的晶体结构均通过粉末x射线衍射确定。xrd图案是使用在45kv/40ma下操作的philips x’pert系统(cu kα辐射,0.1542nm),使用镍kβ过滤器和固态检测器(x'celerator)收集的。使用0.017
°
的步长和0.31s/步的扫描时间进行测量。
[0096]
载体材料给出了预期的衍射图,γ-al2o3中为纯相,tio2中为锐钛矿,zro2中为四方氧化锆。xrd图案如图1所示。添加cs后,载体材料的晶体结构没有变化,显示出相同的衍射图。在45℃下在γ-al2o3和tio2上观察到指示cs2co3的额外衍射峰。碳酸盐据信是由表面cs物质与大气co2反应而形成的。硫化后,碳酸盐及其峰消失,导致形成通过x射线衍射无法检测到的硫氧阴离子。没有其他反射出现。因此得出结论,活性cs物质是xrd无定形的。
[0097]
2.3酸碱性质的表征
[0098]
co和吡啶吸附到纯金属氧化物和所制备的催化剂上是通过红外光谱以透射吸收模式(样品压入自支撑晶片)进行监测,以测量路易斯酸度。在吸附之前,在10ml/分钟的氦气流下,以10℃/分钟的升温斜率将样品加热至360℃。随后,在360℃下将样品在10ml/分钟的在氮气中10%体积%的硫化氢流下硫化0.5小时。为去除物理吸附的硫化氢,在将样品抽真空至10-7
毫巴并冷却至50℃之前,将样品用10ml/分钟的he流再吹扫15分钟。对于吡啶吸
附,将样品池冷却至50℃,并将样品暴露于1毫巴吡啶分压下的吡啶,随后降低吡啶分压。进一步抽真空至10-5
毫巴导致没有吡啶吸附在含cs样品上。因此,在进行抽真空之前,在0.1毫巴下比较不同催化剂的光谱。使用0.96cm/μmol的摩尔积分消光系数计算配位吡啶的浓度,该摩尔积分消光系数是针对1450cm-1
处的特征带测定的。co吸附通过使用液氮将红外单元冷却至-150℃来进行。在5毫巴的co分压下记录光谱。
[0099]
在50℃下吸附甲醇,同时逐步增加甲醇分压(0.1毫巴、0.5毫巴、1毫巴和5毫巴),随后将温度升高至300℃。所有光谱均用nicolet 6700 ftir光谱仪记录(收集64次扫描以获得每个光谱)。所有光谱都经过背景减除,并根据晶片的质量进行归一化。
[0100]
2.4吡啶吸收
[0101]
金属氧化物的酸度是利用通过红外经吸附的吡啶来测量的,图2。在纯γ-al2o3上,在1621、1612、1591、1577、1450和1440cm-1
处观察到八个谱带。1621和1612cm-1
处的谱带分配给与不同酸强度的路易斯酸位点(las)配位结合的吡啶的8a振动模式(波数随酸强度而增加),而1579cm-1
处的谱带分配给8b振动模式。1591cm-1
处的谱带分配给由吡啶与弱酸性表面羟基的相互作用引起的h键吡啶的8a振动模式。1450cm-1
处的信号归因于las上吡啶的9b振动,而1440cm-1
处的谱带再次分配给在羟基上h键合的吡啶。分配给与las配位结合的吡啶的位点(1450cm-1
,1612-1620cm-1
)对抽真空是稳定的,而h键合的吡啶谱带(1440和1593cm-1
)在抽真空后由于其与探针分子的弱相互作用而消失。这与oh基团的释放一致,从而导致3700cm-1
区域周围的负oh谱带减少,因为h键吡啶被解吸(图3)。
[0102]
通过红外,吡啶在zro2和tio2上的吸附得到在1604、1593、1573和1445cm-1
处产生的谱带。1604cm-1
分配给zro2和tio2的与las结合的吡啶的8a振动模式,而1573cm-1
分配给8a振动模式。1593cm-1
分配给由吡啶与弱酸性表面羟基的相互作用引起的h键吡啶的8a振动模式。与γ-氧化铝的情况一样,该信号在抽真空后消失。1445cm-1
处的信号分配给las上吡啶的9b振动。在1450cm-1
处对谱带进行积分,测定金属氧化物上的las浓度为γ-al2o3上的454μmol g-1
、zro2上的220μmol g-1
和tio2上的749μmol g-1
。吡啶信号从γ-al2o3(1450cm-1
)到zro2和tio2(1445cm-1
)的向较低波数的转移表明,前者比后者有较高的路易斯酸强度。
[0103][0104]
表3:所有讨论的金属信号及其分配的总结。
[0105]
在金属氧化物上添加cs,用吸附的吡啶改变了红外光谱(图4)。在中等掺杂的γ-al2o3、cs(10)/γ-al2o3上,不再能检测到分配给与强(1612cm-1
)和弱(1609cm-1
)las配位结合的吡啶的8a振动模式的谱带,以及h键合吡啶的信号。在1583cm-1
处出现了新的谱带,其对应于与弱路易斯酸碱即路易斯酸强度低于在γ-al2o3上测量的那些的cs
+
的配位结合的吡啶的8a振动模式。在zro2和tio2、cs(10)/zro2和cs(10)/tio2上添加cs后,没有观察到分配给载体的las的谱带。与cs/γ-al2o3的情况一样,在1600cm-1
和1583cm-1
处出现了新的谱带,分别对应于cs上吡啶的1+6a和8a泛音振动的振动模式。抽真空后,在所有三个样品上,cs位点上的吡啶信号都消失了,在cs(10)/γ-al2o3和cs(10)/tio2上部分保留。
[0106]
另外的cs、cs(20)/γ-al2o3导致1612cm-1
处谱带的减小。在1600cm-1
处观察到分配给cs上吡啶的1+6a泛音振动的新信号。已经提到的吡啶在cs位点上的1583cm-1 8a振动和吡啶在las和cs上的1573cm-1 8b振动。因此,在γ-al2o3表面逐渐添加cs导致来自γ-al2o3的强las被来自cs的较弱las取代。cs(20)/zro2和cs(20)/tio2催化剂上的吡啶吸附仅导致与cs
+
位点(8a、8b和1+6a)配位结合的吡啶。cs掺杂的zro2和tio2的所有吸附的吡啶物质在真空下解吸;而cs(20)/γ-al2o3上的las的小信号保留。
[0107][0108][0109]
表4:cs负载的金属氧化物上吡啶吸收的分配。
[0110]
用吡啶滴定酸性位点表明具有两种类型las的γ-al2o3中las位点的高度异质性,而tio2和zro2仅提供在两种材料上具有相似强度的一种las,与文献中观察到的那些一致。cs沉积的影响可以合理化为如下:在中等cs负载下,由于较低的sanderson电负性,cs
+
通过直接相互作用改变金属氧化物的表面位点,增加表面碱度。这种直接相互作用是通过表面质子与cs
+
阳离子的交换来完成的。
[0111]
在高cs负载下,表面以cs为主。正如对tio2上的钾所假定的那样,高碱负载导致金属氧化物表面的完全覆盖,从而导致类似于本体碱性材料的表面特性。
[0112]
2.5 co吸收
[0113]
图5显示了通过红外的催化剂的co吸附。不同co谱带的分配见表4。在不同金属氧化物上吸附co时获得了类似的红外谱带。介于2180-2190cm-1
之间的谱带分配给las上的co吸附,而2150cm-1
附近的那些分配给表面羟基。γ-al2o3中的co伸缩振动(2188cm-1
)处于比
zro2(2177cm-1
)和tio2(2181cm-1
)更高的波数,表明co键的更高扰动。这种趋势与用吡啶观察到的趋势相同,意味着γ-al2o3的路易斯酸位点的更高强度。
[0114]
在三种载体上添加10重量%cs导致las上的co伸缩振动降低至较低波数(2138-2136cm-1
),这对应于吸附在cs
+
离子上的co。在cs(10)/γ-al2o3的情况下,在2179cm-1
处出现了另外的谱带,这对应于通过碱性阳离子改变的γ-al2o3载体中的las。没有观察到吸附在oh基团上的co的谱带。在具有20重量%的高cs负载的样品上,仅检测到cs阳离子上的co和物理吸附的co的信号。co没有吸附到cs(20)/zro2样品上。
[0115]
γ-al2o3表面上具有cs的las上的co伸缩振动的红移是由于碱度增加所致,从而降低了sanderson电负性。tio2和zro2的结果与吡啶吸附的结果一致,在10重量%的cs负载下,这些材料上没有可接近的las。与吡啶吸附的情况一样,cs
+
是可用于高cs掺杂材料上的co吸附的唯一物质。通过红外的co吸附与吡啶吸附一致;除了cs(10)/γ-al2o3外,载体的路易斯酸位点在10重量%的cs负载下不存在。
[0116][0117]
表5:纯的和负载cs的金属氧化物上的co吸收的分配。
[0118]
2.6甲醇吸收
[0119]
吸附在金属氧化物及其cs掺杂上的甲醇的红外光谱如图6所示,显示了3000-2750cm-1
区域(烷基(sp3)c-h振动)中的谱带。介于3000-2900cm-1
之间的区域分配给(υ
as
(ch3))的不对称伸缩或其与ch3变形振动(2δs(ch3))的费米共振,而较低的谱带分配给对称伸缩振动(υs(ch3))。对于50℃下的υ
as
和υs,观察到分配给强路易斯酸位点和强路易斯碱位点上的甲醇吸附的红外谱带的不同强度(图6至8)。前一个位点导致形成桥接甲醇盐,称为物质i,其中对于υ
as
(对于γ-al2o3、zro2和tio2为2943、2948和2944cm-1
)和υs(对于γ-al2o3、zro2和tio2为2845、2852和2844cm-1
),红外谱带均处于较高波数。对于υ
as
(对于γ-al2o3、zro2和tio2为2939、2931和2923cm-1
)和υs(对于γ-al2o3、zro2和tio2为2821、2827和2821cm-1
),后一个位点导致形成乙醇化物(o-h基团的解离),称为物质ii。在zro2上,可以看到相对较高浓度的离解甲醇,而对于tio2则进一步增加。在γ-al2o3的情况下,红外单元的加热导致桥接甲醇盐(物质i)的强度增加。在其他两个载体中加热时没有观察到重大变化。甲醇在表面物质上的相对强度直接得出以下结论:金属氧化物相对于更碱性金属氧化物的一般酸性特征按顺序γ-al2o3》zro2~tio2降低。
[0120]
3.根据本发明的负载型催化剂的催化测试
[0121]
甲醇的催化硫醇化在体积为25ml的反应管中进行。在反应之前,在360℃和9巴下将稀释在1 g sic中的125.0mg催化剂(125-250μm)在20ml min-1
h2s流中硫化。与活塞流反应器的空体积(20ml)相比,催化剂的体积几乎可忽略不计。这导致仅0.054h-1
的基于液态甲
醇(ch3oh)的相对低的液时空速(lhsv)。基于完全进料(h2s、ch3oh和n2)的气时空速(ghsv)为150h-1
(基于0℃和1.013巴的标准条件,根据din 1343)。为了确定活化能,用混合有h2s(20ml min-1
)和n2(20ml min-1
)的气态ch3oh(10ml min-1
)流在n2分压为3.6巴、h2s分压为3.6巴且甲醇分压为1.8巴的9巴进料流压力下进行反应。反应管通过夹套借助传热介质加热至300至360℃之间的温度。
[0122]
weisz
–
prater模量的标准计算表明,在所有条件下,对于所有催化剂,它都《1,因此可以得出结论,动力学结果不受内部传质效果的影响。使用配备有hp plot q柱(2.7m,2.0mm内径)的shimadzu gc-2014,使用tcd检测器对产物流进行在线分析。对于用于ch3sh的ch3oh和h2s的0.5级反应,使用积分速率定律计算反应速率常数。为了研究整个转化率范围内的产物分布,调整了停留时间,从而在360℃下将ch3oh的分压保持在2.2巴,将n2和h2s的分压保持在3.3巴。
[0123]
在360℃下确定反应级数。对于h2s的反应级数,甲醇的分压保持恒定在2.2巴,而h2s分压在1.1巴和5.6巴之间变化。为了测量甲醇反应级数,h2s分压设置为4.5巴,ch3oh分压则从0.6毫巴变化到2.2气态ch3oh。调节n2气体流量以补偿体积流量变化,并保持总体积流量恒定在80ml/min。相应地调整每个实验中使用的催化剂量,以确保ch3oh转化率低于10%。用10.0mg催化剂测量铯改性的材料的反应级数,而5.0mg的tio2和zro2及1.0mg的γ-al2o3是足够的。在γ-al2o3的情况下,催化剂以1∶9的比例与sio2物理混合,sio2其在所研究的反应中是惰性的,以避免沟道效应。
[0124]
3.1催化活性
[0125]
甲硫醇(ch3sh)形成的初始速率如图9中所示。观察到tio2有最高甲醇硫醇化速率(0.17-1.4
·
10-6
mol
ch3sh s-1 g
cat-1
),接下来的是γ-al2o3(0.13-9.2
·
10-6
mol
ch3sh s-1 g
cat-1
)和zro2(0.02-0.2
·
10-6
mol
ch3sh s-1 g
cat-1
)。对于cs掺杂的体系,ch3sh形成的速率按cs(10重量%)/γ-al2o3(1.8-8.7
·
10-6
mol
ch3sh s-1 g
cat-1
)》cs(10重量%)/zro2(1.7-7.1
·
10-6
mol
ch3sh s-1 g
cat-1gcat
)》cs(10重量%)/tio2(1.8-6.6
·
10-6
mol
ch3sh s-1 g
cat-1
)的顺序降低。更高的20重量%的cs负载不会导致催化剂活性更高,更确切地,活性对于cs(20重量%)/γ-al2o3(2.0-7.6
·
10-6
mol
ch3sh s-1 g
cat-1
)、cs(20重量%)/zro2(1.7-7.1
·
10-6
mol
ch3sh s-1 g
cat-1
)和cs(20重量%)/tio2(1.8-5.8
·
10-6
mol
ch3sh s-1gcat-1
)略低。虽然用不同金属氧化物的ch3sh形成速率存在一个数量级的差异,但cs体系的活性仅显示出微小的差异。这表明整体活性由表面cs物种决定,其在所有三种金属氧化物载体上似乎都相似。事实上,所有三个体系的ch3sh形成速率都略有下降。
[0126]
测量作为所有三种金属氧化物在360℃下的甲醇转化率函数的甲硫醇、二甲硫醚(dms)和二甲醚(dme)的收率(图10)。在γ-al2o3上,获得ch3sh和(dme)作为初级产物,dme是最高的初级产物,直至60%的甲醇转化率。克服60%的转化率,dme的收率在90%的转化率下降至20%,ch3sh是主要产物。这种行为可以通过dme在催化剂上的重新吸附,发生二次反应形成ch3sh来解释。对zro2观察到类似的结果,ch3sh是主要的初级产物,二甲醚收率低于10%。值得注意的是,在zro2上,在低于10%的转化率下没有形成dme。在γ-al2o3和zro2上,发现二甲硫醚处于较高转化率水平,是ch3sh形成的次级产物。在tio2上没有观察到二甲醚,二甲硫醚是唯一的副产物。在zro2和tio2上进行没有h2s的反应导致形成dme(图10),这暗示zro2和tio2上的反应物之间存在竞争。得到ch3sh的收率按γ-al2o3《zro2《tio2的顺序增加。
测量作为具有10重量%和20重量%cs的所有三种金属氧化物在360℃下的ch3oh转化率的函数的ch3sh、dms和dme的收率(图11)。观察到所有包含cs的体系的总体趋势:ch3sh作为主要产物获得,而唯一产生dme的催化剂是cs(10重量%)/al2o3,其中在360℃下的dme收率为0.3%。主要副产物是dms,其中在360℃下,cs(10重量%)/al2o3上的最大收率为0.7%。在表面上存在cs的情况下不存在dme归因于不存在强的las。这些结果得到了通过红外进行吡啶和co吸附的支持,表明通过cs掺杂显著降低了路易斯酸度。
[0127]
3.2动力学
[0128]
a)甲硫醇的形成
[0129]
甲醇和硫化氢中甲硫醇形成速率对图12中的纯金属氧化物和图13中具有10重量%和20重量%cs的金属氧化物有依赖性。与ch3oh和h2s相关的甲硫醇形成的反应级数如表6所示。
[0130][0131]
表6:针对所有测试体系上的h2s和ch3oh中甲硫醇形成测定的反应级数。
[0132]
在所有金属氧化物上,在ch3sh形成中h2s和ch3oh的0.5的反应级数暗示了两种反应物在双分子langmuir-hinshelwood机制之前的解离。
[0133]
甲硫醇的速率方程为
[0134][0135]
其中a=(1+k
20.5
[ch3oh]
0.5
+k
30.5
[h2s]
0.5
+[ch3sh]
0.5
/k
60.5
+[h2o]
0.5
/k
70.5
)。
[0136]
二甲醚的速率方程为
[0137][0138]
其中b=(1+k
10.5
[ch3oh]
0.5
+[h2o]
0.5
/k
80.5
)。
[0139]
已知硫化氢解离地吸附在金属氧化物的表面上,而甲醇也解离地吸附,从而在表面氧化物的路易斯酸碱对上形成甲醇化物。因此,据信两种底物在相同类型的碱性位点上解离。可以推测,反应级数随着分压的降低会产生底物在表面上竞争吸附的效果。然而,这在金属氧化物的情况下没有观察到。
[0140]
发现甲硫醇形成的表观活化能在γ-al2o3上为约112kj mol-1
,在zro2上为约115kj mol-1
,在tio2上为约107kj mol-1
。这是在金属氧化物的活性位点上形成的甲硫醇的表观活化能,因为不存在cs。
[0141][0142]
表7:针对甲硫醇形成测定的表观活化能.
[0143]
添加cs(10重量%)导致两种反应物的反应级数接近0.5,这暗示与纯金属氧化物所提出的解离反应机制相同。然而,h2s上0.2的反应级数表明cs/tio2和cs/zro2催化剂在h2s的部分覆盖下运转。表观活化能降低到66至78kj mol-1
之间的值。相对于金属氧化物而言,这些催化剂的较低活化能与碱度的增加有关。载体上cs
+
阳离子的存在暗示了其表面羟基的覆盖,类似于氧化铝上就钠和钾所观察到的。在进行吡啶和co吸附期间通过红外确认没有oh谱带证实了这一点。在所有高cs掺杂(20重量%)材料上,获得了与cs 10重量%相似的反应级数值,这也暗示了解离机制。三种高掺杂材料的表观活化位垒在65-59kj mol-1
之间。表观活化能的降低可以通过表面的完全改性来解释。如吡啶吸附所示,在吡啶吸附期间唯一可用的表面物质是cs,这抑制了金属氧化物的化学性质并充当非常弱的las。此外,强路易斯酸位点的缺失仅导致表面甲醇化物的形成,如在高cs掺杂催化剂上的甲醇吸附期间通过红外观察到的那样。
[0144]
针对所有的纯金属氧化物,确定用于dme形成的甲醇和硫化氢的反应级数(表8和图14)。在γ-al2o上,用于二甲醚形成的甲醇中的反应级数为1.5,h2s中的反应级数为0。h2s的零级表明h2s不与dme形成位点上的甲醇竞争。甲醇的1.5反应级数可由甲醇部分覆盖催化剂表面来解释。在γ-al2o3上,与h2s相比,甲醇的吸附似乎是有利的,导致dmf的形成和h2s的零级。发现在zro2和tio2上,用于dme形成(不存在h2s)的反应级数为0.7。据信与γ-al2o相比,在这些材料上甲醇的表面覆盖率更高。与其他两种材料相比,γ-al2o3上较低的表观活化能归因于较高的路易斯酸强度,如co和吡啶吸附所示,从而促进ch3oh的co键的断裂。
[0145][0146]
表8:在甲醇和硫化氢中的反应和用于二甲醚形成的表观活化能。
[0147]
3.3催化活性
[0148]
对于纯金属氧化物催化剂,γ-al2o3对于甲硫醇形成具有最低的选择性,其中在300至320℃的温度范围内,二甲醚是主要产物(其中s
dme,300℃
=71.2%且s
dme,320℃
=63.4%)。随着温度升高,对二甲醚的选择性降低到s
dme,340℃
=49.8%且最终s
dme,360℃
=28.6%。随着二甲醚选择性的降低,甲硫醇选择性从300℃下的28.5%增加到360℃下的71.2%。对二甲硫醚形成的选择性小于5%。
[0149]
在纯zro2上,形成甲硫醇的选择性在300℃下为53.1%,在360℃下为60%。同样,
主要副产物是二甲醚,然而选择性则从300℃下的46.7%降低到360℃下的36.6%。与γ-al2o3一样,对二甲硫醚的选择性小于5%。
[0150]
在所有的纯金属氧化物中,tio2提供对甲硫醇形成最高的选择性:在300℃下为96%,其中在更高的温度下选择性降低(s
dme,360℃
=79.2%)。与其他两种金属氧化物相比,对二甲醚的选择性随着温度的升高从s
dme,300℃
=4.0%增加至s
dme,360℃
=16.6%)。二甲硫醚的生产选择性小于4%。
[0151]
与纯金属氧化物催化剂相比,所有cs负载催化剂都显示出ch3sh选择性的显著增加。对于γ-al2o3上10重量%的cs,ch3sh形成的选择性增加到s
ch3sh,300℃
=99.7%至s
ch3sh,360℃
=98.3%的范围。对副产物的选择性随温度的升高而增加:以s
dme,300℃
=0.1%至s
dme,360℃
=0.5%的选择性形成dme,并且以s
dms,300℃
=0.2%至s
dms,360℃
=1.2%的选择性形成dms。cs负载进一步增加到20重量%也将ch3sh选择性提高到高达s
ch3sh,300℃
=99.9%至s
ch3sh,360℃
=99.1%的选择性。在这种情况下,发现的唯一副产物是选择性为s
dms,300℃
=0.1%至s
dms,360℃
=0.9%的dms。
[0152]
对于zro2系催化剂,对ch3sh的选择性增加到s
ch3sh,300℃
=99.9%至s
ch3sh,360℃
=99.1%的范围。再次,发现的唯一副产物是选择性为s
dms,300℃
=0.1%至s
dms,360℃
=0.9%的dms。将cs负载增加至20重量%导致介于s
ch3sh,300℃
=99.9%和s
ch3sh,360℃
=99.4%之间的甚至更高的选择性。
[0153]
对于tio2系催化剂发现了类似的结果:在10重量%cs的情况下,对ch3sh的选择性增加至介于s
ch3sh,300℃
=99.9%至s
ch3sh,360℃
=99.4%之间的范围。再次,发现的唯一副产物是选择性为s
dms,300℃
=0.1%至s
dms,360℃
=0.6%的二甲硫醚。将cs负载增加至20重量%再次导致介于s
ch3sh,300℃
=99.9%和s
ch3sh,360℃
=99.5%之间的甚至更高的选择性。
[0154][0155]
表9:所制备催化剂的产物选择性的总结(n.d.=不可检测)
[0156]
4.对比例
[0157]
比较例是用在γ-al2o3上包含cs2ws4的催化剂进行的。所述催化剂通过两步初始润湿浸渍法而制备。首先,将5.0gγ-al2o3(类似于sph 509 axens,晶粒尺寸为150-250μm)用溶解在1.6ml h2o中的0.64g乙酸铯(sigma aldrich,≥99.99%)浸渍。将样品在室温下干燥过夜,以得到cs/al2o3。接下来,按如下合成cs2ws4/al2o3体系:通过沉淀形成cs2ws4晶体,将350mg(nh4)2ws4在20ml h2o中的溶液和325mg cs2co3在20ml h2o中的溶液混合。形成黄色沉淀。过滤这些固体,用冰冷的水和1-丙醇洗涤。由于cs2ws4的溶解度低,因此将450mg的这些溶解在150ml水中。然后将2g cs/al2o3添加到溶液中。通过连续旋转蒸发来除去水,在固体样品上沉淀cs2ws4晶体。将样品在室温下干燥过夜。干燥后,将样品在455℃下(升温速率为5℃/min)煅烧4小时。所制备的催化剂具有5.1重量%的钨含量、20.6重量%的铯含量、0.20cm3g-1
的孔体积和141m2g-1
的bet表面积,两者均按以上所述测量。使用配备有质谱仪(qme 200,pfeiffer vacuum)的流动设备,利用脉冲技术进行吸附,接下来是h2s的温度程序脱附。将催化剂的样品装入石英反应器并在以6ml/min流速的4.2体积%h2s/he下在360℃下原位活化2小时。对于h2s吸附,温度设置为360℃,将样品在吸附前用he吹扫1小时。每30分钟引入4.4体积%的在he中的h2s的脉冲(5.0μmol/min的h2s)。所吸附的气体总浓度计算为每个脉冲吸收的总和。
[0158]
在与实施例3相同的反应条件和相同的反应管下测试由此获得的催化剂。在测试
之前,通过在20ml/min流速的h2s中在360℃下处理2小时来活化催化剂。
[0159]
在300℃、320℃、340℃和360℃的温度下,meoh的转化率,ch3sh、dme和dms的收率,以及对ch3sh、dme和dms的选择性总结在下表10中。
[0160][0161]
表10:对比例的结果的总结
[0162]
ch3sh形成的初始速率如图15所示。在300℃的温度下观察到甲醇硫醇化的最高速率(1.34
·
10-6
mol
ch3sh s-1 g
cat-1
),在更高的温度下具有以下速率:在360℃的温度下具有最低的速率(6.38
·
10-6
mol
ch3sh s-1 g
cat-1
)。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1