碱性阳离子富集和水电解提供CO2矿化和全球范围碳管理
碱性阳离子富集和水电解提供co2矿化和全球范围碳管理
1.相关申请的交叉引用
2.本技术要求2019年6月14日提交的美国临时专利申请第62/861,848号的权益,该临时专利申请的全部内容以引用方式并入本文。
3.关于由联邦政府资助研究或开发的声明
4.本发明是根据由美国能源部授予的授权号de
‑
fe0029825、de
‑
fe0031718、de
‑
fe0031705在政府支持下完成的。政府对本发明享有一定的权利。
背景技术:
5.需要对现有的大气二氧化碳和持续的二氧化碳排放进行管理,以减轻全球平均温度的升高并减少气候变化的影响。为了真正地实现这一点,在下个世纪,每年必须从大气中除去约10
‑
20千兆吨(gt)的co2,因此需要可以大规模实施的碳管理策略。
6.在处理流体状态(例如,气体、液体或超临界)的co2方面调整比较碳捕集和储存。这种流体状态处理在用于co2的通路、过程和处置途径方面增加了限制和复杂性,包括昂贵且耗能的分离、用于压缩的大能量需求以及具有泄漏可能性的加压容器的高成本。
7.将从点源或大气中捕集的co2注入地质构造(包括:(枯竭)油气藏、不可采煤层和咸水含水层)可在北美洲封存高达22,000gt的co2。虽然理论容量非常大,但实际上,在50年的注入中,防止岩石破裂或现有断层的再活化所需的压力限制和/或残余烃的存在导致每年约700mt的更适度的储存容量。尽管预期地质封存场所的可能容量超过足以适应co2排放的当前(和未来)水平的容量,但是co2迁移和泄漏的风险以及注入过程的管理和验证需要随时间对井、地下和地面进行大量监测。此外,基于碳捕集、封存和储存(ccss)的碳管理的传统方法受以下项影响:(i)与从空气或烟道气料流中分离co2的熵以及随后满足从固体或液体底物中解吸co2的焓的需要相关的热力学罚分,以及(ii)对用于将co2输送到地质封存场所的巨大物流和运输基础设施(例如,管道)的需要。具体地,在常规的基于吸附/解吸的co2捕集中,能量消耗与co2从气体混合物中的分离相关,包括体系的熵的降低、允许co2浓度达到足以用于管道运输的等级的解吸步骤、以及随后的地质封存。综上所述,虽然技术挑战仍然存在,并且正在逐步得到解决,但ccss的实际实现很大程度上取决于支持性政策,该政策可以在全球范围内为待定的最佳实践和限时监测授权、降低风险,在极限情况下使ccss项目的开发人员免受损害。
8.除了地质封存和储存之外,土地使用、农业实践、海洋地质工程和co2向建筑材料的化学转化的变化提供了替代的大规模途径,构成用于确保碳管理(例如,减排和大气碳去除)的方法的组合。虽然在负(co2/碳)排放技术(net)的开发中已取得一些进展,但还需要更多的实质性“指数”进步来以成本有效/可行的方式实现必要的co2去除速率和耐久的碳储存。
9.针对这一背景,需要开发在本公开中描述的实施方案。
技术实现要素:
10.本公开的一些实施方案包括通过以下从含水料流或气态料流中去除二氧化碳的方法:使当存在时包含二氧化碳的气态料流与包含能够形成不溶性碳酸盐的离子的水溶液接触;使包含二氧化碳的水溶液与诱导其碱化的电活性网格接触,从而迫使一种或多种碳酸盐固体从溶液中沉淀;以及从溶液中或从碳酸盐固体可能沉积的网格的表面去除所沉淀的碳酸盐固体。在一些实施方案中,存在气态料流。在一些实施方案中,气态料流包含介于0.04体积%至100体积%之间的co2。在一些实施方案中,气态流体为大气空气。在一些实施方案中,气态流体为从天然气发电厂、燃煤发电厂、铁厂、钢厂、水泥厂、乙醇厂和化工制造厂排放的烟道气。在一些实施方案中,水溶液包含与气态料流平衡的一定量的溶解二氧化碳。在一些实施方案中,水溶液与气态料流处于热平衡。在一些实施方案中,水溶液不与气态料流处于热平衡。在一些实施方案中,不存在气态料流。在一些实施方案中,能够形成不溶性碳酸盐的离子包含包括以下中的一种或多种的离子:ca、mg、ba、sr、fe、zn、pb、cd、mn、ni、co、cu和al。在一些实施方案中,水溶液具有约1,000ppm或更高的nacl浓度。在一些实施方案中,水溶液具有约30,000ppm或更高的nacl浓度。在一些实施方案中,水溶液包含海水。在一些实施方案中,电活性网格包括网状阴极,该网状阴极包含金属或非金属组合物。在一些实施方案中,方法利用每吨矿化的二氧化碳约2.5mwh或更小的端对端能量强度。在一些实施方案中,水溶液包含缓冲至大气丰度的一定量的溶解二氧化碳。在一些实施方案中,电活性网格在水溶液中在约2μm至20000μm的电活性网格内原位产生增加的碱性条件。在一些实施方案中,碱化条件为9或更大的ph。在一些实施方案中,电活性网格由金属或碳基网格组成。在一些实施方案中,电活性网格包含不锈钢、氧化钛、碳纳米管、聚合物和/或石墨,或者这些材料的其他混合组合物。在一些实施方案中,电活性网格包括直径在约0.1μm至约10000μm范围内的孔。在一些实施方案中,诱导碳酸盐固体的沉淀包括在溶液中旋转由电活性网格组成的圆筒,同时施加吸力以将溶液抽吸到网格的外表面上。在一些实施方案中,溶液为盐水溶液。在一些实施方案中,溶液为含碱金属的溶液。在一些实施方案中,诱导碳酸盐固体的沉淀包括诱导至少一种具有ca、mg、ba、sr、fe、zn、pb、cd、mn、ni、co、cu或al的碳酸盐的沉淀。
11.本公开的一些实施方案包括流通式电解反应器,该流通式电解反应器包括:与包括电活性网格的旋转圆筒流体连接的吸入装置,以及用于从表面或溶液中分离固体的刮擦装置和/或基于液体喷雾的装置。在一些实施方案中,反应器还装有包含二氧化碳、ca离子和mg离子的水溶液。在一些实施方案中,电活性网格能够通过从包含二氧化碳和能够形成不溶性碳酸盐的离子的水溶液中沉淀碳酸盐固体来诱导溶解无机碳的去除。在一些实施方案中,电活性网格包含金属或碳基网格。在一些实施方案中,电活性网格包含不锈钢、氧化钛、碳纳米管、聚合物和/或石墨,或者这些材料的混合组合物。在一些实施方案中,反应器包括多个电活性网格。在一些实施方案中,多个电活性网格被布置成一系列平行的平面单元或平行的圆柱形单元。在一些实施方案中,反应器与脱盐装置流体连通。
附图说明
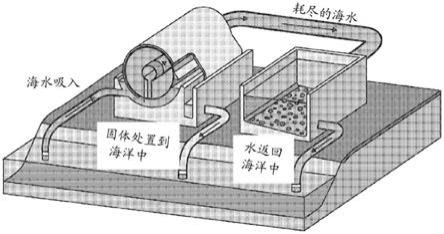
12.图1示出了二氧化碳矿化和处置方法的实施方案。加热元件和扩散器是任选部件。
13.图2(a)示出了代表性计算,其证明了对于如下组成的溶液,方解石沉淀的极限受
ca、co2或碱度的影响:[ca]=10mm,[cl]=20mm,[co2]=10mm(约30%co2;300000ppm),ph=4.16(虚曲线)。最大caco3产率为10mmol/kg水。如果[ca]降低至3mm或5mm(深蓝色曲线)([cl]为6mm或10mm),[co2]=10mm,ph=4.17,则最大caco3产率降低至3mm和5mm。类似地,[co2]降低至3mm(浅蓝色曲线)(约9%co2,ph=4.42)或5mm(约15%co2,ph=4.31)([ca]=10mm,[cl]=20mm)会降低最大caco3产率。有意思的是,对于相等的caco3产率,降低[ca]会导致naoh消耗增加,而降低[co2]会降低naoh消耗。图2(b)对以下两种情况进行了比较:(i)初始pco2为5%,对应于1.73mm co2(粗曲线),并允许随沉淀而降低,以及(ii)溶液的pco2保持恒定(例如,通过与17300ppm的气态co2料流连续平衡)在5%(细曲线)。如图2(a)所示,在(i)中,caco3沉淀通过添加naoh而被迅速诱导,并且受到总溶解co2的限制。在(ii)中连续供应co2能够使caco3以约7mm的产率沉淀(受[ca]限制)。(c)在如henry定律规定的不同pco2水平(体积%)下与co2平衡的溶液的方解石沉淀的naoh消耗量。
[0014]
图3示出了在气相浓度范围内与co2气态料流平衡的溶液的作为ph的函数的总溶解碳含量。总溶解co2等于[h2co3*]+[hco3‑
]+[co
32
‑
]。作为参考,0.04%表示空气中co2的浓度。为了比较,示出了空气中的co2含量。
[0015]
图4示出了对于参考海水组成,使用具有llnl.dat数据库的phreeqc的代表性平衡计算,如millero等人(deep sea research part i:oceanographic research papers 2008 55(1),50
‑
72)给出。以1:2的摩尔比同时添加co2和naoh导致方解石和菱镁矿的沉淀,每kg水至多约55mmol菱镁矿和约10mmol方解石。大气压下的co2饱和浓度为约34mm。在工程化方法中,可通过在碳酸盐沉淀过程中简单地鼓入空气来维持co2平衡(参见图2b、图2c)。
[0016]
图5示出了对于使用aspen模拟的基于胺的方法,作为浓度的函数的co2捕集和压缩的能量要求(红色实曲线)。从混合物中分离co2并从1atm压缩至15mpa所需的热力学最小能量(红色虚曲线)基于(分离)混合气相co2的熵进行计算。还示出了在生产的热力学最小能量需求下氯碱生产的naoh(蓝色虚线)的co2矿化(蓝色实线)的能量成本。由nacl生产naoh的理论能量需求获自thiel等人(acs sustainable chem.eng.2017,5(12),11147
‑
11162)。使用koh/k2co3方法进行直接空气捕集和压缩(dacc;品红色三角形)和集成苛性胺(绿色圆圈)的能量成本分别获自keith等人(joule 2018,2(8),1573
‑
1594,climatic change 2006,74(1),17
‑
45)。垂直的灰色线表示空气、天然气发电厂和燃煤发电厂以及水泥厂烟道气中的co2浓度。阴影区域表示以下co2缓解策略的代表性能量成本范围:(红色)捕集和压缩(范围:每吨co
2 0.1mwh(在热力学最小值下)至4.5mwh;具体取决于浓度),(蓝色)电解合成的naoh的化学计量添加(范围:每吨co
2 1.26mwh(在热力学最小值下)至4.5mwh),以及(黄色)电解沉淀方法(单步碳封存和储存)(范围:每吨co
2 0.07mwh(在热力学最小值下)至2.3mwh;与浓度无关)。
[0017]
图6(a)示出了在图6(b)的膜阴极上局部生成oh
‑
作为诱导碳酸盐沉淀的手段的概念图。使用转鼓过滤解决方案去除沉积在膜表面上/附近的物质。图6(c)示出了如使用comsol使用时步自适应、三角形网格元件(173.21μm2的网格开口)和周期性边界条件所模拟的电极化后1s的不同过电位的电解质的ph。假设水的击穿电位为0v
rhe
(rhe:可逆氢电极)(参见rsc adv.2019,9(54),31563
‑
31571)。该模拟考虑由浸入过量电解质(0.1m nacl)中的304l不锈钢构成的平面电极(100mm2),其中对于析氢反应(her),tafel关系[η=0.172+log(i/i0),其中η为过电位(v),i为电流密度(a/m2),并且i0为交换电流密
度(1.04x10
‑7a/m2)]表示:ph(t)=14+log[{(10
η
‑
1.2)/0.172]
)t}/9.6485+10
‑7],其中ph为在1mm厚的区域内的邻近盐水电解质中产生的平均ph,η为过电位(v),并且t为时间(s)。例如,需要约0.5v的过电位以在膜表面产生10的ph,在该ph下,溶液中的所有无机碳以co
32
‑
阴离子的形式形成。预期地,增加电极的表面积(例如,使用网格)或其电化学活性将降低诱导近表面碱化所需的过电位。
[0018]
图7示出了用于实现co2矿化和处置的单步碳封存和储存的概念图。
[0019]
图8(a)示出了304l不锈钢在0.1m nacl溶液中的阴极极化曲线。虚线表示析氢反应(her)的tafel拟合:η=0.172+log(i/i0),其中η为过电位(v),i为电流密度(a/m2),并且i0为交换电流密度(1.04x10
‑7a/cm2)],表明在时间t(s)处1mm厚的区域中的电解质的ph(t)=14+log[{(10
[(η
‑
1.2)/0.172]
)t}/9.6485+10
‑7]。图8(b)示出了环形反应器(插图)的作为过电位(例如,电池电位与水击穿电位之差1.23v)的函数模拟的阳极与阴极之间的平衡ph。通过oh
‑
和h
+
离子的产生和传质以及通过酸碱中和(oh
‑
+h
+
→
h2o)来控制ph。
[0020]
图9(a)示出了具有平面几何结构的电解沉淀器的剖视图。图9(b)示出了具有圆柱形几何结构的电解沉淀器的剖视图。在两种构型中,酸化的(c,ca,mg)耗尽的海水出口进料可用于硅酸盐风化以提高流出物的碱度和ph。
[0021]
图10(a)示出了使用comsol模拟的具有圆柱形孔(100μm,半径)的1mm厚网格的几何结构,例如图10(b)中的阴极的剖面。图10(a)示出了对于1mm/s的流速,不同电流密度的孔溶液的ph,并且图10(c)示出了对于1ma/cm2的电流密度,不同平均流速的孔溶液的ph。
[0022]
图11(a)示出了碳纳米管
‑
聚乙烯醇(cnt
‑
pva)复合膜,其显示出表面形态和约125nm的孔径。图11(b)示出了在流动反应器中施加1v(电池)电位后暴露于10mm cacl2溶液的阳极活性碳(石墨)电极表面(孔径20μm)的光学图像(fov为约1mm
×
1mm),显示出通过热重量分析鉴定为caco3的沉淀物。与入口相比,出口处的较低的[ca]进一步证明了溶解的ca
2+
的去除。
具体实施方式
[0023]
本公开的某些实施方案涉及固定co2的方法。
[0024]
根据一些实施方案的一个方面,一种方法包括:将二氧化碳引入溶液中;以及诱导碳酸盐固体从溶液中沉淀,其中诱导碳酸盐固体的沉淀包括对溶液进行水电解。在一些实施方案中,经由气体扩散器将二氧化碳引入溶液中。在一些实施方案中,溶液经由与大气平衡而包含(溶解)二氧化碳。在一些实施方案中,对溶液进行水电解包括增大进料溶液的ph。在一些实施方案中,对溶液进行水电解包括生成氢氧根离子。在一些实施方案中,诱导碳酸盐固体的沉淀包括在溶液中旋转膜鼓,同时施加吸力以将溶液抽吸到膜鼓的表面上。在一些实施方案中,溶液为盐水溶液。在一些实施方案中,溶液为含碱金属的溶液。在一些实施方案中,诱导碳酸盐固体的沉淀包括诱导碳酸钙或碳酸镁或其他碳酸盐(例如,碳酸钡)或其他相关固体中的至少一种的沉淀。在一些实施方案中,方法还包括富集溶液中的碱金属阳离子。
[0025]
在根据一些实施方案的另一方面,一种方法包括:将二氧化碳引入溶液中;以及诱导碳酸盐固体从溶液中沉淀,其中诱导碳酸盐固体的沉淀包括在溶液中旋转膜鼓,同时施
加吸力以将溶液抽吸到膜鼓的表面上。在一些实施方案中,经由气体扩散器将二氧化碳引入溶液中。在一些实施方案中,诱导碳酸盐固体的沉淀包括对溶液进行水电解。在一些实施方案中,对溶液进行水电解包括增大进料溶液的ph。在一些实施方案中,对溶液进行水电解包括生成氢氧根离子。在一些实施方案中,溶液为盐水溶液。在一些实施方案中,溶液为含碱金属的溶液。在一些实施方案中,诱导碳酸盐固体的沉淀包括诱导碳酸钙或碳酸镁或其他碳酸盐(例如,碳酸钡)或其他相关固体中的至少一种的沉淀。在一些实施方案中,方法还包括富集溶液中的碱金属阳离子。
[0026]
在根据一些实施方案的另一方面,一种通过以下从含水料流或气态料流中去除二氧化碳的方法:使当存在时包含二氧化碳的气态料流与包含能够形成不溶性碳酸盐的离子的水溶液接触;使包含二氧化碳的水溶液与诱导其碱化的电活性网格接触,从而迫使碳酸盐固体从溶液中沉淀并通过电解去除溶解无机碳;以及从溶液中或从碳酸盐固体可能沉积的网格的表面去除所沉淀的碳酸盐固体。在一些实施方案中,存在气态料流。在一些实施方案中,气态料流包含介于约0.04体积%至100体积%之间的co2(例如,约0.04体积%、0.1体积%、0.2体积%、0.5体积%、1体积%、2体积%、3体积%、4体积%、5体积%、10体积%、20体积%、30体积%、40体积%、50体积%、60体积%、70体积%、80体积%、90体积%、95体积%、98体积%、99体积%、99.9体积%的co2,以及其间的范围)。在一些实施方案中,气态流体为大气空气。在一些实施方案中,气态流体为从天然气发电厂和燃煤发电厂、钢厂和铁厂、水泥厂、乙醇厂和化工制造厂等排放的烟道气。在一些实施方案中,水溶液包含与气态料流平衡的一定量的溶解二氧化碳。在一些实施方案中,水溶液例如在5℃≤t≤100℃的温度下与气态料流处于热平衡。在一些实施方案中,水溶液例如在5℃≤t≤100℃的温度下不与气态料流处于热平衡。在一些实施方案中,不存在气态料流。在一些实施方案中,能够形成不溶性碳酸盐的离子包含包括以下中的一种或多种的离子:ca、mg、ba、sr、fe、zn、pb、cd、mn、ni、co、cu和al。在一些实施方案中,水溶液具有约1,000ppm或更高的nacl浓度。在一些实施方案中,水溶液具有约30,000ppm或更高的nacl浓度。在一些实施方案中,水溶液具有约1000ppm、2000ppm、3000ppm、4000ppm、5000ppm、10000ppm、20000ppm、30000ppm、40000ppm、50000ppm、60000ppm以及其间的范围的nacl浓度。在一些实施方案中,水溶液包含海水或微咸水或盐水。在一些实施方案中,电活性网格包括网状阴极,该网状阴极包含金属或非金属组合物。在一些实施方案中,方法利用每吨矿化的二氧化碳约2.5mwh或更小的端对端能量强度。在一些实施方案中,水溶液包含缓冲至大气丰度的一定量的溶解二氧化碳。在一些实施方案中,电活性网格在水溶液中在约2μm至20000μm的电活性网格内原位产生增加的碱性条件。在一些实施方案中,碱化条件为9或更大的ph(例如,约9、10、11、12、13、14以及其间的范围的ph)。在一些实施方案中,电活性网格包括金属或碳基网格。在一些实施方案中,电活性网格包含不锈钢、氧化钛、碳纳米管、聚合物和/或石墨,或者这些材料的其他混合组合物(例如,金属/聚合物、金属/非金属、金属/陶瓷)。在一些实施方案中,电活性网格包括直径在约0.1μm至约10000μm范围内(例如,约10μm、50μm、100μm、200μm、300μm、400μm、500μm、1000μm、1500μm、2000μm、3000μm、4000μm、5000μm、6000μm、7000μm、8000μm、9000μm或10000μm)的孔。在一些实施方案中,诱导碳酸盐固体的沉淀包括在溶液中旋转由电活性网格组成的圆筒,同时施加吸力以将溶液抽吸到网格的外表面上。在一些实施方案中,溶液为含碱金属的溶液。在一些实施方案中,诱导碳酸盐固体的沉淀包括诱导至少一种
具有ca、mg、ba、sr、fe、zn、pb、cd、mn、ni、co、cu和al的碳酸盐的沉淀。
[0027]
在根据一些实施方案的另一方面,一种流通式电解反应器包括:与包括电活性网格的旋转圆筒流体连接的吸入装置,以及用于从网格表面/溶液中分离固体的刮擦装置和/或基于液体喷雾的装置。在一些实施方案中,流通式电解反应器还包括包含二氧化碳、ca离子和mg离子的水溶液。在一些实施方案中,电活性网格能够通过从包含二氧化碳和能够形成不溶性碳酸盐的离子的水溶液中沉淀碳酸盐固体来诱导溶解无机碳的去除。在一些实施方案中,电活性网格包含金属或碳基网格。在一些实施方案中,电活性网格包含不锈钢、氧化钛、碳纳米管、聚合物和/或石墨,或者其他混合组合物。在一些实施方案中,反应器包括多个电活性网格。在一些实施方案中,多个电活性网格被布置成一系列平行的平面单元或平行的圆柱形单元。在一些实施方案中,反应器与脱盐装置流体连通。
[0028]
co2可以固定在稳定的矿物碳酸盐内。策略的基础是在含水介质中从源自液体和固体料流的气态co2与ca
2+
和/或mg
2+
离子(或能够形成不溶性碳酸盐的其他离子,诸如ba、sr、fe、zn等)的组合沉淀固体碳酸钙(caco3)、碳酸镁(mgco3)以及它们的变体。然后,矿化碳可在地球表面被处置或被排出到海洋中。大的碳储存容量、极小的环境影响和低的晚期co2释放风险支持该方案作为长期gt级co2废物管理的主要途径的可行性。
[0029]
碱性co2矿化方法可通过将强碱诸如naoh添加到与海水类似的接近中性的含ca和mg的进料溶液(例如,包含约10毫摩尔每升(mm)cacl2和约55mm mgcl2的纯水)中来实现,如下所述。进料溶液也可由液体料流诸如富含碱金属的地下水、工业废水、脱盐盐水等构成。碱性阳离子富集构成任选的预处理阶段以增加ca、mg和其他碱性阳离子的水性浓度。富集可通过过滤、电容浓缩或它们的组合来实现。然后,可使用曝气池(诸如在废水处理的活性污泥工艺中使用的那些)来获得有效的混合和co2平衡以产生富含co2的水。可将碱(例如,naoh)共混到诸如用于水处理的凝聚和絮凝过程中的富含co2的水中,导致caco3和mgco3的沉淀。沉淀物可通过沉降分离,并且排出的固体可进一步脱水以进行填埋或类似于脱盐设备中的盐水处置那样排放到海洋中。
[0030]
碳酸盐矿化的热力学和动力学可通过在膜表面上诱导的局部ph和温度偏移来进一步增强。代替添加消耗性试剂,先进的co2矿化方法涉及二氧化碳矿化和处置设备,其通过水电解(经由水电解器)生成氢氧根离子(oh
‑
)并且升高现场的温度(经由加热元件)。更偏碱性的溶液增加了碳酸盐沉淀的驱动力,并且通过水电解生成oh
‑
增大了液体的ph以促进碳酸盐沉淀。类似地,较高的温度增加了碳酸盐沉淀的驱动力,并且加热元件的使用升高了液体的温度以促进碳酸盐沉淀。任选地经由气体扩散器和压缩机将co2注入或以其他方式引入液体中;或者可以与其气态气氛平衡的水平存在于液体中。如图1所示,设备的设计为在置于池内的富含co2和含ca/mg的液体(例如,海水或其他盐水溶液)中旋转的旋转真空膜鼓的形式。膜鼓包括膜(例如,网格或筛网形式的金属膜)作为围绕中心管道的过滤介质,通过该中心管道施加吸力(或负压)。当膜鼓旋转通过液体时,可在膜的表面形成碳酸盐。真空泵连接到中心管道并施加吸力以将液体抽吸到膜表面上并通过膜表面,其中ca
2+
和mg
2+
与co
32
‑
一起沉淀为碳酸盐。ca
‑
/mg
‑
贫乏的滤液流到膜鼓的内部并被泵出。碳酸盐固体粘附到膜鼓的外部,然后膜鼓经过刀片以从膜去除固体,从而当鼓旋转回到液体中时再生膜表面以用于随后的碳酸化。这种先进的方法还提供了与脱盐设备集成/集成在脱盐设备内的方法的可能性,同时解决了co2导致的气候变化的问题和膜脱盐设备中的结垢问题(由膜表
面上的ca
2+
和mg
2+
化合物的积聚引起)。
[0031]
实施例
[0032]
碳酸钙例如方解石的沉淀由下式给出:
[0033][0034]
其中k
sp
为溶度积(也称为平衡常数)并且等于ca
2+
和co
32
‑
在平衡时的水活度的乘积。在方解石的沉淀过程中,hco3‑
或co
32
‑
(两者均通过co2在水中的形态(speciation)而形成)可吸附并结合在生长表面上。caco3的k
sp
随温度降低,使得方解石饱和溶液的温度从25℃升高至90℃导致方解石沉淀,产率为约300μmol/kg水。描述co2‑
h2o体系的形态反应和解离常数写为:
[0035][0036][0037]
其中h2co3*表示总co2(水溶液)和h2co
30
。溶解co2的分配由bjerrum图公开。一般来讲,co
32
‑
阴离子的活度取决于ph(例如,在水中,co
32
‑
是ph>10.33时的主要碳物质),方解石沉淀的程度也是如此。较高的盐度使k1和k2偏移至较大的值,因此hco3‑
和co
32
‑
离子占主导时的ph偏移至较小的值。矿物沉淀的热力学驱动力由饱和度ω=iap/k
sp
给出,其中iap为离子活度积;例如,对于方解石,这是溶液中的ca
2+
和co
32
‑
的活度的乘积。这与含有二价金属阳离子的典型天然水(例如,地下水、海水、采出水)相关,因为它们的接近中性的ph需要提供补充的碱度以诱导碳酸盐沉淀。因为ca和mg是天然水和通常工业用水(例如,采出水、热蒸发盐水等)中最丰富的二价阳离子;这些水表示为cacl2溶液,因为在很大程度上,cl
‑
提供对这些体系中的阳离子的电荷补偿。
[0038]
添加naoh提高溶液的ph和ω
方解石
,最终导致方解石沉淀。对于固定初始co2浓度(pco2)的溶液,方解石沉淀受到ca、co2的丰度或碱度(ph)的限制。这些情况在图2(a)中示出,该图考虑了在各种浓度的ca、co2或碱度下添加co2的cacl2溶液。尽管较低酸性的溶液(例如,具有较低的co2浓度)需要较少量的naoh来引发方解石沉淀,但在这种情况下受co2限制的最大caco3产率可因此较低(图2a中的浅蓝色曲线)。另一方面,溶液中过量的co2可使ca成为限制反应物。在其中naoh:caco3和h2o:caco3摩尔比均被最小化的情况下,ca和co2摩尔浓度大致相等,并且添加naoh(表示碱度)直到实现最大caco3产率(图2a中的虚曲线)。naoh的添加导致na
+
与co
32
‑
的络合;因此,{co
32
‑
}和{ca
2+
}之间达到等效(其中括号{}表示活度)的co2与ca之间的摩尔比略高于1。消耗的naoh与作为caco3封存的co2之间的摩尔比最小为2(例如,对于反应[2]和[3]中的每一者,其提供一摩尔oh
‑
)。
[0039]
对于以低co2浓度为特征的液体料流,空气可以相对于co2消耗速率鼓泡通过含水进料。为了说明这一点,在图2(b)中比较了两种情况:在一种情况中,初始co2浓度固定且溶解碳逐渐耗尽,在另一种情况中,溶液的co2浓度通过与以固定pco2为特征的气态料流平衡而保持恒定。在第一种情况下,caco3沉淀通过添加naoh而被迅速诱导并且受到总溶解co2的限制,而在第二种情况下,co2的补充使方解石沉淀,直到ca耗尽。在第二种情况下,由于过量的酸性,例如co2,沉淀最初被抑制。为完整起见,用于改变pco2水平的co2平衡/补充的不同情况示于图2(c)中。在极限情况下,碳酸盐产率仅取决于进料的[ca]丰度并且不随co2浓度变化。可溶于水的co2的量由其ph和盐度以及温度依赖性亨利定律常数控制。对于给定溶
液,增大溶液ph(ph>6)会增加总溶解碳(图3)。这是由于co2(水溶液)的ph依赖性形态为hco3‑
和co
32
‑
,这降低了co2(水溶液)的浓度,允许根据亨利定律进一步溶解co2(气体)。正是由于这个原因,相对于ph>6的淡水,海水中阳离子
‑
碳酸盐和阳离子
‑
碳酸氢盐络合物的形成增加了其碳储存容量。
[0040]
碳酸盐沉淀反应通过时间尺度来表征。在25℃和1atm下的充分混合的条件(例如,没有传质限制)下,由反应[4]描述的平衡在t=5.0x10
‑
11
s内发生。
[0041][0042]
如反应[5]
‑
[7]所述,含水物质h2co3、hco3‑
和co
32
‑
在10
‑2s内达到平衡。
[0043][0044][0045][0046]
然而,相对于ca
2+
的平衡(即,反应[8]和[9])仅在103s内达到。
[0047][0048][0049]
在碱性溶液(ph>10)中,通过与oh
‑
反应形成hco3‑
的co2溶剂化的替代途径甚至比与h2o反应(k=6.6x10
‑4m
‑1s
‑1)更快(k=8.5x103m
‑1s
‑1)。在类似于海水(≥0.5m nacl)的浓溶液中的方解石沉淀速率表示约3.2x106m s
‑1的沉淀速率常数;其产率与图2(c)一致。该速率常数通过使用以下形式的方程拟合实验(方解石沉淀)数据而得到:r=k(ω
‑
1)
n
,其中r为以m s
‑1为单位的沉淀速率,k为以m s
‑1为单位的速率常数,ω为相对于方解石的饱和度,并且n为反应级数。因此,在具有低传质阻力的充分混合的体系中,与ca
2+
的反应(例如,caco3沉淀)是限速的。这允许分析此后的co2矿化的质量和能量平衡。
[0050]
确立基线。一般来讲,上述分析表明碱度促进碳酸盐沉淀。因此,最重要的是检验向接近中性的含ca和mg的溶液中添加强碱诸如naoh的典型方法(图4)。作为参考,应当注意的是,来自点源排放的co2浓度对于天然气发电厂为约3%,对于燃煤发电厂和钢铁厂为15%,对于水泥厂为20%,并且对于氨、乙醇和氢气厂为>90%(体积%)。另一方面,大气co2浓度为约0.04%,而通过胺洗涤捕集的co2的纯度可大于99%。因此,本分析包括0.04%、5%、20%和100%的参考浓度。在基线情况下设想的co2矿化通过模拟水处理过程来建模。首先,如果使用除空气之外的co2源,可以简单地使用类似于活性污泥处理中使用的曝气池来实现与盐水的有效混合和co2平衡。此后,如在凝聚和絮凝过程中一样,naoh可混合到富含co2的水中,导致caco3和mgco3的沉淀。然后沉淀物通过沉降从溶液中分离,并且排出的固体可使用压带机脱水或类似于脱盐中的盐水处置那样排放到海洋中。
[0051]
确定用于碳酸盐矿化的钙和镁充足性。ca和mg以超过充足的量用于满足全球范围的碳管理的需求。例如,每年除去10gt co2需要9.1gt ca或5.5gt mg,相当于全球海洋(“海水”)中所含总ca的0.0017%和总mg的0.00032%。另选地,尽管在小得多的水平下,钙和镁可以源自:(a)可含有超过1,000mg/l总溶解固体(tds)的地下盐水,其在2015年提取率达到每年32亿m3,对应于0.6mt ca和0.3mt mg(在美国使用微咸水中的典型ca和mg浓度);(b)全球以每年500亿m3的速率生产的脱盐盐水每年可提供额外的0.04gt ca和0.1gt mg;以及
(c)每年仅在美国产生的2.23gt采出水(假设平均ca浓度为5,000mg/l)每年可提供额外的0.01gt ca。虽然由金属、合金和水泥的制造以及由煤燃烧产生的碱性副产物富含ca和mg,但推测它们的风化每年固定不超过0.3gt co2。总之,海水仍然是矿化方法中最可行且最丰富的二价金属离子源。
[0052]
从化学计量来看,1mol co2转化为1mol caco3需要2mol naoh(参见图2a和图4)。因此,10gt的co2的矿化最少需要大约18gt naoh。然而,全球生产的naoh相对较少;在2016年为大约70mt,但仅基于其na含量,可使用海水合成3.2x107gt naoh。为满足这一需求,将需要6,000个大型氯碱厂,每个厂每年生产3mt naoh;但这是一个不可行的提议。对于海水足量的naoh用量具有将每kg水2.86g co2转化为mgco3和caco3的潜力(图4)。因此,为了每年封存10gt co2,每年需要处理约3500gt水;这一数量类似于我们全球的年取水量(约4000gt)。另一方面,在美国,每年在超过14,700个处理工厂中处理47gt废水。如果每年单个co2削减设施处理2000mt(例如,大型废水处理厂的规模)的海水,那么全球将需要建造1760个这样的工厂,每个工厂每年供应10mt naoh。因为碳酸盐产率受到进料中的二价阳离子含量的限制,所以在进料料流中富集ca和mg浓度(例如,使用可以选择性地分离二价阳离子的膜)将允许处理较少量的水。显然,这样的预处理只能在似乎不可行的大量能量损耗的同时才能实现;尽管将导致碳酸盐产率的增加。
[0053]
能量强度分析。为了与地质ccss策略进行比较,可以估计使用海水作为二价阳离子源并使用naoh作为化学计量添加剂的矿化方法的能量需求。与地质ccss不同,基于海水矿化的co2削减不需要co2捕集步骤。因此,基线过程的能量需求(尽管实际上不可行)基于以下需求:水处理和加工,以及naoh生产。水处理和加工包括:(a)海水吸入,其需要约1.3kwh/t co2矿化,(b)化学分散,其需要2.8
‑
7.7kwh/t co2矿化,以及(c)沉降,其需要0.175
‑
0.35kwh/t co2矿化。因此,总体上,假设海水进料,水加工和处理可消耗约5kwh/t矿化co2。通过氯碱法合成naoh需要2.5mwh/t naoh。因此,直接co2矿化(使用海水作为二价离子和(溶解)co2的来源)的能量需求估计为大约4.5mwh/t co2(图5)。因此,对于目前同类最佳的氯碱生产的naoh,由每mwh约70美元的工业用途电价估计的co2去除成本为315美元/t co2。通过使碱性固体(例如,其溶解在溶液中产生碱金属(ca
2+
、mg
2+
)和oh
‑
)碳酸化,可稍微降低对naoh的需要。用于这种直接碳酸化的能量投入包括预处理成本,包括研磨和在一些情况下的热活化、(碳酸盐)产物处置、以及泵和混合器的操作,并且为大约0.5mwh/t co2(全部包括在内)。不幸的是,工业碱性固体的这种直接碳酸化预期每年提供不超过0.3gt的co2削减。可通过双极膜电渗析实现能量消耗较少的naoh生产途径。即使可以0.7mwh/t naoh的理论最小能量需求生产naoh(具有可用于增强硅酸盐溶解的副产物hcl的45%naoh),基于矿化的co2管理的成本也将至少与1.26mwh/t co2的能量强度相关(图5中的蓝色虚线水平线);例如,对应于每t使用得自海水的二价阳离子转化为固体碳酸盐的co2不低于90美元的成本。
[0054]
传统ccss途径的能量强度通过考虑基于单乙醇胺(mea)的工艺来估计,该工艺由吸收器、汽提器、冷却器和四级压缩机组成,使用aspen和erntl热力学性质方法(参见图5)。在本文中,假设co2耗尽的气体从吸收器的顶部提取,而富含co2的溶剂料流从底部提取。为了释放co2,加热富含co2的胺以实现解吸焓。塔顶冷凝器向塔提供回流液体料流,并且
将富含co2的气体纯化至接近100%co2。从汽提器释放的接近纯的co2料流被压缩并传输以用于地质储存。考虑到具有3%co2的入口料流(例如,对应于从天然气发电厂排放的烟道气),对于基于胺的co2捕集(用于胺再生的热负荷为1.3mwh/吨)以及对于回收的co2料流(0.2mwh/吨)从大气压加压至14mpa下的管道规格,估计约1.5mwh/t co2的能量强度(参见图5;红色实曲线)。随着co2在入口料流中的浓度增加至约12%,该能量强度降低至0.8mwh/吨co2(对于燃煤发电厂;0.6mwh用于碳捕集,并且0.2mwh用于压缩每吨co2);其后保持恒定。然而,在入口co2浓度低于3%co2时,基于胺的工艺的能量强度急剧增加。与空气接触的胺的低容量(约0.25mol/mol)将需要大于5.0mwh/吨co2的再沸器负荷,以实现小于0.05mol/mol的工作量。这表明对于其中co2进料料流异常稀释的情况,诸如在从大气中去除co2时或大气平衡的水,替代的矿化方法的独特优点在于操作上能量消耗较少。
[0055]
单步碳封存和储存(scs2):上述讨论表明,基于矿化的co2管理的能量消耗主要与为该方法提供碱度的需要相关。理想的碳封存方法将不需要消耗性化学品投入,这导致制造、运输、处理和储存成本。在理想情况下,该方法可使用零碳电子(例如,来自光伏)来提供动力。无化学品投入的单步碳封存和储存(scs2)方法示于图6和图7中。在此,含有溶解co2(与空气平衡)的水(例如,海水)流过多孔金属膜/阴极。阴极电位的施加导致水电解和膜/水界面处局部升高的oh
‑
浓度,这促进碳酸根阴离子和金属阳离子的快速结合,同时使传输限制最小化,并提供用于异相成核的基底。具体地,使电解质流过膜的孔使得所有离子种类(oh
‑
、co
32
‑
和me
2+
)的扩散长度尺度(至孔半径)最小化,同时为金属碳酸盐的成核和生长提供表面位点。有限元分析(fea)表明,在合理的过电位(≈0.5v;参见图6c)下,确实可以在电活性膜(“电极”)表面附近产生超碱性条件。实际上,200μm的电极/膜表面内的电解质体积(例如,远大于针对此类应用设想的电活性膜的孔径)在电极化的1s内经历超碱性条件,足以诱导碳酸盐沉淀至图5所示的限度。作为参考,在本文中,将304l不锈钢电极表示为平面片材;但在实际应用中设想了具有大约20μm的开口的粗网格。该分析证实施加温和的电位能快速生成碳酸盐沉淀所需的碱度。尽管该分析忽略了导致对流混合的电迁移和气体逸出,但其提供了诱导快速碱化进而诱导碳酸盐沉淀所需的过电位的下限估计。
[0056]
基于电化学oh
‑
生成的电解矿化方法的实际能量需求可以基于以79%效率运行的现有技术的电解器来估计(例如,假设对于化学计量的析氢反应的热力学需求为39.4kwh/kg,50kwh的电力生成1kg的h2)。经由水的电解产生的1kg h2产生1000摩尔的oh
‑
离子,在化学计量基础上,对于2.3mwh/吨co2的能量强度,该离子可封存22kg co2。如果考虑共同产生的氢气的热值,假设转化效率(例如,用于燃烧氢气和产生电)为大约60%,例如类似于天然气燃烧,则该方法产生1.2mwh/吨矿化co2的能量强度。该分析考虑其中2mol oh
‑
将1mol co2矿化为碳酸钙(caco3)的化学计量。根据该基准,每吨矿化co2将生成45kg低压h
2(g)
。预期这种氢气提供大约3美元/kg的商业价值,使得每吨矿化co2可实现大约135美元的成本补偿。另一方面,如果使用氢燃料电池(hfc)将所产生的低压氢气转化为电力,则可实现大约80%的转化效率,使得将产生0.84mwh/吨矿化co2的能量强度。对于以90%效率运行的电解器,能量强度进一步降低至1.9mwh/吨co2(无h2回收;133美元/吨co2)和0.38mwh/吨co2(使用hfc以90%效率回收和转化h2;27美元/吨co2)。这些值限制了scs2的实际能量强度(参见图5中的黄色区域)。总之,该分析表明:(i)直接电解矿化可以在co2的环境浓度下实现效率是典型的基于氯碱的naoh生产和基于胺溶剂的方法几乎两倍的碳去除(例如,保守地,小于
2.3mwh/吨相较于大于4mwh/吨),并且(ii)如果考虑氢气副产物的能量益处并/或使用零碳能量投入;在每种情况下,scs2方法为开拓性的、真正的负排放技术(net)提供了基础。
[0057]
使用电活性膜进行矿化以局部产生碱度的显著优点在于,由于ph升高、过饱和(ω;参见图4和图6c)以及由焦耳加热引起的在膜表面产生的温度升高(高达60℃,在溶液中),它增强了沉淀动力学(成核和生长)。该方法的电解性质需要在阴极条件下机械且化学稳定的导电(例如,金属或复合材料)膜。由于金属碳酸盐的形成,预期这种膜表面在操作期间会结垢。预期scs2方法的工程设计将匹配电解沉淀反应器内的雷诺数、佩克莱数和达姆科勒数;使得(反应物)传质和化学沉淀动力学彼此对应地发生。虽然这不可能导致如本文所考虑的粗网格结构中的显著通量衰减,但是矿物碳酸盐的绝缘性质确实可以损害电流密度(例如,从而增加所需的过电位)和能量效率。因此,可能需要循环地反转所施加的电位以在膜表面附近阳极生成o2和h
+
,从而去除沉积物。然而,阳极条件可导致快速腐蚀,特别是在使用铁基膜的情况下。另一种方法涉及沉积碳酸盐的物理磨损,其方式类似于用于连续清洁转鼓过滤器的方式(例如,参见图6b、图7)。在这些系统中,用刀片连续刮擦膜的表面,该刀片除去积聚的固体并使膜表面再次暴露。毫无疑问,scs2概念以用于碳削减的高能量强度为特征;此概念:(a)比大多数其他直接空气捕集(dac)方法更有效,(b)允许直接使用无碳电力;例如尤其是在过量时,并且(c)确保端对端co2削减。此外,本文设想的电解反应器可以简单地与现有的和未来的脱盐设备模块化集成,而不要求新的氯碱设备的构造,从而允许在产生可饮用水和可用作燃料的氢气的同时去除和封存co2。scs2方法的附加有益效果是生成软化水,该软化水是脱盐设备的优良进料。目前,考虑到海水反渗透(swro)要求2
‑
2.5kwh/m3,并且预处理步骤(例如,实质上包括水软化)消耗0.3
‑
1.0kwh/m3海水,海水脱盐的能量成本可估计为3.5kwh/t水。将基于co2矿化的预处理和swro脱盐相组合可导致比这两种方法单独操作的总能量消耗低9%的能量使用。
[0058]
然而,重要的是,即使需要阳离子补充/ph调节,也可通过利用在阳极处随之发生的酸度产生在电化学系统中容易地实现。具体地,在工程化系统中,废弃海水料流的电解(再)碱化可通过使用在scs2过程中共同产生的酸度以增强风化的方式溶解镁铁质和超镁铁质岩石以及包括煤燃烧和金属加工残留物在内的工业固体来进行。值得注意的是,该分析表明,与固体相比,单位碱度以水溶液形式储存的co2的量更大。而每摩尔作为碳酸盐固体储存的c需要2摩尔oh
‑
,每摩尔作为溶解离子储存的c仅需要1.2摩尔oh
‑
。结果,将ph从8增大至9(例如,1μm oh
‑
至10μm oh
‑
)可使每kg水多溶解33mmol co2(图3)。因此,排出的水可被设计成具有比提取的海水更大的ph(例如,更高的[ca]和/或[mg]);以提供进一步增强的碳削减有益效果。因此,去除大气co2的策略应谨慎地与海水(再)碱化相组合,以增强其由海洋
‑
大气平衡驱动的co2容量。
[0059]
碳酸盐固体的命运。co2在矿物碳酸盐内的截留可以快速发生,并且提供稳定且持久的储存,同时消除任何封存后释放的风险。假定化学计量和方解石的沉淀,从大气中去除10gt co2(溶解于海水中)可导致每年产生约20gt固体。这些固体中的一些可以替代跨越建筑材料(骨料)和特殊应用的全球石灰石市场。在美国,生产的碎石的68%是复合碳酸盐岩;约1gt产量用于建筑和作为水泥生产原料。不能利用的固体可经由现有的固体废物管理策略进行处置。2016年,全球城市固体废物产生以及工业、农业、建筑和拆除废物总量为约25gt。在美国,固体废物的填埋成本为约45美元/t,10gt碳酸盐固体的填埋处置每年需要约
6.8km3(68亿m3)的空间。固体可以储存在废弃矿山中,而不是建造新的填埋场。2017年,全球开采了53gt金属和非金属矿石材料、15gt化石燃料和24gt生物质。然而,异地储存需要运输固体,成本为约0.03美元/m3/km。更现实的是,特别是如果使用海水作为碱性源,沉淀物可能会重新沉积在海洋中(例如,以脱盐盐水的形式;其中由于海洋中方解石和菱镁矿过饱和,这些固体可以保持稳定并且不反应),或者用于土地开垦和防侵蚀目的。
[0060]
根据2006年修订和执行的london protocol,除了附录1的“反向清单”中列出的可能可接受的废物外,禁止海上倾倒。碳酸钙和碳酸镁可被认为是“惰性地质材料”,允许在海洋中进行处置,它们在海洋中可以保持稳定,因为近地表海水在这两相方面是过饱和的。如果溶解的ca和mg取自除海水以外的来源(例如,含盐地下水),则沉淀的ca和mg碳酸盐可用于缓冲由大气co2吸收或直接co2注入、通过添加和溶解石灰石而引起的海洋ph降低。回到土地开垦,加利福尼亚南部海岸线迁移的简单模型近似于海平面上升1m时后退约30m。假设大陆架的平均深度为50m,在延伸4500km的海岸线上产生20gt的固体可以逆转这种影响;约为佛罗里达州墨西哥湾沿岸长度的一半。通过co2矿化衍生的固体创造新的土地不仅可以解决未来的co2排放问题,还可能逆转气候变化最显著的影响之一。可以解决因海平面上升而导致陆地和栖息地消失的危机,同时提供永久且不需要持续监测的co2储存解决方案。对经由scs2方法补贴co2管理的机制、特别是相关资本成本的深入分析超出了本研究的范围。尽管如此,美国和加利福尼亚州的低碳燃料标准(lcfs)中最近的45q税收抵免通过隐含地将co2隐性定价在每吨35
‑
180美元之间来激励碳减排。这种激励为实现和增强全球范围的co2减排和减少提供了重要且潜在的必备途径。
[0061]
这种解决co2问题的scs2途径不同于传统的碳捕集和地质封存策略。与常规的基于吸附剂的co2捕集方法不同,在这些常规方法中,大量的能量消耗与以下所需的能量消耗相关:(1)从气体混合物中分离通常为稀浓度(小于15体积%)的co2,以及(2)对于co2解吸,scs2方法依赖于电解促进的碳酸盐矿物沉淀,该沉淀在可以完全使用可再生能源操作的过程中进行。然而,需要弥合在无碳电力的供应中的主要差距以实现实际的可行性。最后,通过稳定固体碳酸盐,该方法消除了监测和验证co2封存和储存的需要,同时提高并确保了co2储存的持久性。鉴于碳储存需要持续数千年,电解海水co2矿化的组合只有在与加速的硅酸盐/碳酸盐风化相组合时才能为我们提供可行的、环境友好的并且可能比传统的地质封存更可接受的解决全球碳危机的方法;特别是在短期到中期(5至10年)部署net时。
[0062]
在一些实施方案中,scs2方法包括电活性网格组合物以及它们在“可拆卸和可堆叠”流动反应器中的集成,以在专为直接空气捕集(dac)定制的策略中实现可缩放的co2减排,而不需要任何化学计量的试剂或添加剂(参见图7)。在本文中,与空气平衡的包含溶解碱性阳离子和溶解co2的水(例如,采出水、地下水或海水)流过多孔导电网格/阴极。阴极电位的施加导致水的电解和网格/水界面处局部升高的oh
‑
浓度,这通过使传输约束最小化并提供用于异相成核的基底来促进碳酸根阴离子和金属阳离子(例如,ca
2+
、mg
2+
)的快速结合(图7)。该假设有两个基本原理:(1)阴极表面上oh
‑
的电化学生成和金属碳酸盐的n&g是快速反应。因此,晶体生长速率受到离子向生长核的传输的限制。沿着多孔网格/电极电化学生成oh
‑
同时使电解质流过网格的孔最大限度地减少了所有离子物质(oh
‑
、co
32
‑
、ca
2+
和mg
2+
)的扩散长度尺度(至孔半径),同时为金属碳酸盐的n&g提供表面位点,从而提高反应速率。(2)由于晶体生长的能量势垒较低(例如,对于caco3,5kj/mol对比12.5kj/mol),相对于
均相成核,非均相成核是有利的。因此,提供ph最高的表面(例如,网格/电极)促进碳酸盐的沉淀,同时封存大气来源的co2。
[0063]
为了支持该方法,实验数据和有限元分析(fea)表明,在合理的过电位(≈0.5v)下,在电活性网格/阴极表面附近(例如,在200μm内)容易产生超碱性条件(ph>10)。虽然在网格表面上形成碳酸盐可限制电子转移反应,但scs2方法可包括物理方法(例如,刮擦和/或水擦洗),以类似于商业转鼓过滤器中使用的方式除去沉淀物并更新网格/阴极表面。沉淀物可以类似于脱盐盐水的方式作为悬浮固体收集和/或丢弃(图7)。通过溶解碱性岩石和工业固体(煤燃烧残留物)对废弃海水料流进行电解(再)碱化可允许以增强风化的方式从大气中吸收更多的co2。此外,与用于dac的现有吸附剂和膜相比,本文可开发的电活性网格结构具有优异的性能(例如,基于每单位捕集、矿化或削减的co2的能量)。
[0064]
该方法利用了相对于通过大气
‑
海水平衡调节的空气在水中高得多的co2浓度。当前平均ph为8.1的海水含有的co2是同等体积空气的150倍(图3),因此显著减少了待处理流体的体积。尽管海水和空气的相对密度大于该浓度系数,但是泵送水比空气更有效,并且去除相同量的co2所需要处理的水量比空气少。此外,scs2方法利用了碳酸盐从碱性溶液中沉淀的有利热力学。沉淀的热力学驱动力由饱和状态ω=iap/k
sp
给出,其中iap为离子活度积,并且k
sp
为溶度积。对于方解石,这是溶液中和平衡状态下ca
2+
和co
32
‑
的活度的乘积。吉布斯自由能差是根据下式的溶液组成的函数:δg=rt inω,其中r为气体常数,并且t为温度。co2(气体)(
‑
394.3kj/mol)、co
32
‑
(
‑
527.8kj/mol)和caco3方解石(
‑
1129.1kj/mol)的标准生成吉布斯自由能的比较表明,碳酸盐沉淀(“co2矿化”)在热力学上呈下降趋势。电化学碱化提高溶液的ph和ω
方解石
,从而确保方解石(和/或菱镁矿或其他碳酸盐)沉淀(图4和图6)。与碳酸盐在碱性溶液中沉淀相关的反应是快速的。在25℃和1atm(1巴)的充分混合条件下,由条件下,由描述的平衡在t=5.0x10
‑
11
s内发生。含水物质h2co3、hco3‑
和co
32
‑
如下所述:如下所述:和在10
‑2s内达到平衡。在碱性溶液(ph>10)中,通过与oh
‑
反应形成hco3‑
的co2溶剂化的替代途径甚至比与h2o反应(k=6.6x10
‑4m
‑1s
‑1)更快(k=8.5x103m
‑1s
‑1)。高盐水(≥0.5m nacl)中方解石沉淀速率的数据表明沉淀速率常数为3.2x106m s
‑1,产率与图4一致,其通过将实验沉淀数据拟合为以下而得到:r
p
=k(ω
‑
1)
n
,其中r
p
为沉淀速率,k为速率常数,ω为饱和指数,并且n为反应级数。因此,在具有低传质阻力的充分混合的体系中,caco3沉淀是限速的。净反应为:ca
2+
+co2+2oh
‑
→
caco3+h2o以及mg
2+
+co2+2oh
‑
→
mgco3+h2o。按照这些化学计量,1mol co2被1mol含水ca
2+
或mg
2+
捕集,并且需要2mol oh
‑
以产生1mol caco0或mgco3。对于典型的海水,在阳离子受限的环境(相关的边界条件)中,每处理1000g水就矿化2.86g co2(图4)。
[0065]
为清楚起见,模拟了缩放反应器中的ph分布。该模拟考虑了如下的电极反应:(1)在阳极处:析氧反应(oer):2h2o
→
o2+4h
+
+4e
‑
;(2)在阴极处,(2a)氧还原反应(orr):o2+h2o+4e
‑
→
4oh
‑
,以及(2b)析氢反应(her):2h2o+2e
‑→
h2+2oh
‑
。
[0066]
304l不锈钢的电化学行为示于图8(a)中。溶液中的溶解氧在负过电位下促进orr(相对于her),并且在0至0.4v的过电位下达到4x10
‑7a/cm2的扩散限制电流。这种限制电流可通过her克服(例如,水击穿),其电流遵循与所施加的过电位的tafel关系。orr可在阴极
产生高达ph 10的局部碱度,而her促进更高的ph产生;尽管处于较高的电池电位。
[0067]
scs2方法由一个主单元操作组成,如图9所示。在本文中,将在大气条件(约23℃,1巴总压力和约400ppm co2)下用来自空气的co2饱和的富含ca和mg的水(例如,海水、采出水、地下水)带入电解流动反应器中。
[0068]
作为非限制性实例公开了两种构型,使用以下任一种:(a)平面电极(图9(a))或(b)管状电极(图9(b))。在(a)中,网状阴极放置在由非导电材料构成的矩形壳体的中心,从而形成两个室(图9(a))。阳极插入其中一个室的壁附近。在(b)中,阳极和阴极被径向放置在非导电管内,类似于图4所示的构型(图9(b))。海水(105mg co2当量c/l,参见图3)流过由下述网格/阴极
‑
阳极系统组成的沉淀反应器。可使用具有95%
‑
100%oer效率的氧选择性材料(例如,mno2)作为阳极上的涂层以允许使用单一电解质(海水),从而抑制由海水电解引起的阳极处cl
2(g)
的析出。在反应器内,阴极电位的施加使阴极/水界面处的[oh
‑
]升高,导致沉淀。这显示在电活性网格的孔内ph分布的模拟中(图10)。近中性海水进入孔并变得越来越呈碱性直至ph 12(图10(a)),达到取决于电流密度(图10(b))和流速(图10(c))的程度。
[0069]
利用基于膜的水处理系统的技术优点以及在环境条件下海水中co2的量显著高于空气中的量,同时利用热力学有利的矿化反应,电活性网格组合物和流动反应器使海水介导的dac成为可能。由于膜结垢不会影响scs2方法,并且事实上是其目标,因此其允许简单的固体机械去除和/或循环极性反转作为膜再生的手段。
[0070]
scs2方法比现有的直接空气捕集(dac)方法更加节能。首先,通过考虑由吸收器、汽提器、冷却器和四级压缩机组成并使用aspen的基于单乙醇胺(mea)的方法来估计传统碳捕集和储存(ccs)的能量强度。在入口co2浓度<3体积%co2时,能量需求急剧上升,在0.04%时外推至>3mwh/t co2,这主要是因为在低负载下从溶剂解吸中co2所需的热能增加。scs2的能量需求主要与水电解相关。以79%效率运行的现有技术的电解器(例如,假设her的热力学需求为39.4kwh/kg,50kwh的电力生成1kg h2)产生1000摩尔的oh
‑
离子,在化学计量基础上,对于2.3mwh/吨co2的能量强度,该离子可矿化22kg co2。对于以90%效率运行的电解器,能量强度降低至1.9mwh/吨co2。因此,图4中方法的预期功率要求(每天2kg co2)为约0.2kw(每天约4.6kwh)。泵送水还需要能量:(i)穿过网格(对于40μm网格开口,≈10psi;1.2kwh/t co2)以及(ii)对抗重力(例如,1米的总动力压头;1.3kwh/t co2)。
[0071]
表1.基于电活性网格的co2去除系统的状态点数据
[0072]
[0073][0074]
在本文的一些实施方案中公开的方法在功能上类似于基于膜的dac方法。然而,去除是基于电诱导的化学反应,而不是大小或电荷排斥。诸如(1)处理能力、(2)能量强度和(3)碳酸盐单程产率的度量可提供类似于传统的基于膜的方法所寻求的相关信息。数据表明电解沉淀反应是快速的;k≈3.2x106m s
‑1。因此,产率受到存在的阳离子量的限制。对于ca、mg限制的反应,60%和100%的转化率得到“测量的”和“预计的”度量。
[0075]
在一些实施方案中,使用氢燃料电池(hfc)将所产生的低压氢气转化为电力,可实现大约80%的转化效率,使得将产生0.84mwh/t矿化co2的净能量强度。
[0076]
(a)电活性网格材料和(b)网格集成到其中的流动反应器允许水的碱化和促进超快沉淀的可行性已被证明能用于处理含铬水,其中沿着格表面/阴极的ph摆动使得cr(oh)3能够在膜/水界面处快速积聚(图11)。网格包括:(a)基于316l不锈钢(ss)网格或穿孔网格的基线网格,(b)非金属碳基网格(碳纳米管(cnt)/聚合物/剥离石墨复合物),以及(c)具有局部烧结钛膜(马格涅利相烧结ti4o7材料)的ss网格。选择后两种网格组成以提供:高电导率和在海水中的稳定性,特别是在可腐蚀铁基材料的阳极条件下(可能需要定期清洁网格)。所选材料具有:低成本、易加工性,这使得能够容易地制造许多形状因子,包括多孔(例如,具有μm至mm范围的开口的粗网格)结构。例如,多孔ti4o7材料易于通过tio2粉末的烧结和热还原进行制造。还有可能:(i)应用刮刀加工以快速产生跨越100s in2的碳电极网格材料,并且(ii)通过将cnt的渗透网络气刷到多孔聚合物载体上,然后与聚合物例如聚乙烯醇(pva)交联来制造大型cnt基膜(参见图11)。这些复合材料是稳定的和导电的,具有nm至mm的可变孔径。
[0077]
可使用适于在盐水溶液中引起ph摆动的一系列金属和碳基网格/电极。具体地,多孔几何结构(例如,网格或非织造垫)的使用由以下构成:不锈钢(ss)(烧结网格为约12美元/m2,或约0.05美元/g)、马格涅利相烧结ti4o7材料(由tio2合成,约0.10
‑
0.20美元美分/g)、以及碳纳米管(cnt)/聚合物/剥离石墨(eg)复合材料(例如,cnt成本为约3
‑
30美元/g,并且由石墨合成的eg为约0.10美元/g)。可使用具有各种孔隙率(15%
‑
40%)和孔径(0.1μm
‑
100μm,对应于压降<15psi)的网格材料(<5cm x 5cm)(较小的孔允许较高的ph和较低的过电位,但需要较大的驱动力来推动水通过)。对于ss,可使用市售的网格材料(例如,由304和316l ss制成)和孔径在37μm(400目)和1μm之间的烧结金属片(对于烧结ss板)。为了制备ti4o7涂覆的网格,可以购买tio2粉末,将其浇铸在凝胶中并在1050℃的流动空气下烧结,然后在1050℃的流动h2气体下还原;这是产生亚化学计量的ti4o7的条件。替代合成方法是经
由溶胶
‑
凝胶法和真空碳热法的组合。可通过将cnt/eg悬浮液喷涂到多孔聚四氟乙烯和不锈钢载体上来制造碳基网格,并使用pva交联。网格表面形态和孔径可使用扫描电镜(sem)来评估;网格粗糙度可使用原子力显微镜(afm)来评估;并且孔径可使用sem来评估。网格的组成可使用能量色散x射线光谱(eds)、傅立叶变换红外光谱(ftir)和使用rietveld分析的定量x射线衍射来确定。可使用四点电导率探针、循环伏安法、电化学阻抗谱和电化学显微镜来表征整体电化学特性。网格的长期稳定性可在1至200ma/cm2的电流密度下在死端过滤池中连续操作>168小时来评估,其中进料料流(海水)被加压以流过可用作阴极的网格。mno2涂覆的ti棒可用作阳极。
[0078]
可以对不同的网格材料进行原位原子力显微镜检查(afm)以优化电流密度和(水)通量,用于经由海水电解沉淀碳酸盐。这可以确定使碱化动力学与热力学预测一致的最佳表现网格组成;同时使固体沉淀物形成最大化。配备有流体单元和温度控制、稳压器和允许高速成像的光热探针激发模块的电化学afm可用于筛选分析。
[0079]
可以监测网格表面的电流密度和碳酸盐过度生长层的形貌,同时将各种过电位(0.0v至2.0v)施加到安装在包含模拟海水的流体单元中的1cm x 1cm x≤0.25cm网格样品上。流体单元以分开的液体和气体交换端口为特征。通过使用可编程注射泵交换流体单元内的溶液或气体,可以实时控制网格暴露的水性环境(例如,在施加电位和数据收集期间)。例如,为了补充溶解的ca
2+
和mg
2+
(例如,其通过caco3沉淀从溶液中提取),模拟海水以与其从溶液中耗尽的速率相匹配的流速通过密封单元交换。另一方面,为了在不补充阳离子的情况下补充溶解的大气co2,空气可流过单元。碳酸盐生长的动力学(例如,速率、形态)可通过在数秒至数小时的时间段内收集时间序列图像来评估。可通过测量纵横比、厚度和表面覆盖率来跟踪沉淀物的形态,该纵横比、厚度和表面覆盖率可通过在网格表面处引起电阻损耗/焦耳加热来影响电解沉淀的进展。还可以评估固定溶液组成和pco2随时间的沉淀物生长速率的变化。溶液ω可通过对表面处的ph演变和气/液交换速率进行建模来估计。因此,可以识别使生长速率最大化的电解条件(例如,施加的电位、流速、ω)。可以选择例如由于电阻损耗而使得在最低过电位下碳酸盐沉淀的产率和速率最高并且碳酸盐生长速率随时间降低的网格。在监测表面形貌/电流密度的同时,可以在10s的极化反转内测试所选网格的循环性能。
[0080]
定义
[0081]
除非上下文另外明确说明,否则如本文所用,单数术语“一个”、“一种”和“该”可包括复数指代物。因此,例如,除非上下文另外明确说明,否则对对象的引用可包括多个对象。
[0082]
如本文所用,术语“组”是指一个或多个对象的集合。因此,例如,一组对象可以包括单个对象或多个对象。一组中的各个对象也可被称为该组的成员。一组中的各个对象可为相同的或不同的。在一些实例中,一组中的各个对象可以共享一个或多个共同特征。
[0083]
如本文所用,术语“连接”是指操作联接或链接。连接的对象可以直接地彼此联接,也可以间接地彼此联接,诸如经由一个或多个其他对象。
[0084]
如本文所用,术语“基本上”和“约”用于描述和说明小的变化。当与事件或情况结合使用时,该术语可以指事件或情况精确发生的实例以及事件或情况近似发生的实例。当与数值结合使用时,该术语可以指小于或等于该数值的
±
10%的变化范围,诸如小于或等于
±
5%、小于或等于
±
4%、小于或等于
±
3%、小于或等于
±
2%、小于或等于
±
1%、小于
或等于
±
0.5%、小于或等于
±
0.1%、或小于或等于
±
0.05%。
[0085]
另外,量、比率和其他数值有时在本文中以范围型式呈现。应当理解,这样的范围型式是为了方便和简洁而使用,并且应当灵活地理解为包括明确指定为范围界限的数值,而且还包括涵盖在该范围内的所有单个数值或子范围,如同明确指定了每个数值和子范围。例如,在约1至约200范围内的比率应当理解为包括明确列举的约1和约200的界限,但也包括单独的比率诸如约2、约3和约4,以及子范围诸如约10至约50、约20至约100等。
[0086]
附加实施方案
[0087]
e1.一种方法,包括:
[0088]
将二氧化碳引入溶液中;以及
[0089]
诱导碳酸盐固体从溶液中沉淀,其中诱导碳酸盐固体的沉淀包括对溶液进行水电解。
[0090]
e2.如e1所述的方法,其中通过气体扩散器将二氧化碳引入溶液中,或者溶液可包含与其环境处于平衡水平的二氧化碳。
[0091]
e3.如e1
‑
e2中任一项所述的方法,其中对溶液进行水电解包括增大进料溶液的ph。
[0092]
e4.如e1
‑
e3中任一项所述的方法,其中对溶液进行水电解包括产生氢氧根离子。
[0093]
e5.如e1
‑
e4中任一项所述的方法,其中诱导碳酸盐固体的沉淀包括在溶液中旋转膜鼓,同时施加吸力以将溶液抽吸到膜鼓的表面上。
[0094]
如e1
‑
e5中任一项所述的方法,其中溶液为盐水溶液。
[0095]
e7.如e1
‑
e6中任一项所述的方法,其中溶液为含碱金属的溶液。
[0096]
e8.如e1
‑
e7中任一项所述的方法,其中诱导碳酸盐固体的沉淀包括诱导碳酸钙或碳酸镁中的至少一种的沉淀。
[0097]
e9.一种方法,包括:
[0098]
将二氧化碳引入溶液中;以及
[0099]
诱导碳酸盐固体从溶液中沉淀,其中诱导碳酸盐固体的沉淀包括在溶液中旋转膜鼓,同时施加吸力以将溶液抽吸到膜鼓的表面上。
[0100]
e10.如e9所述的方法,其中经由气体扩散器将二氧化碳引入溶液中。
[0101]
e11.如e9
‑
e10中任一项所述的方法,其中诱导碳酸盐固体的沉淀包括对溶液进行水电解。
[0102]
e12.如e9
‑
e11中任一项所述的方法,其中溶液为盐水溶液。
[0103]
e13.如e9
‑
e12中任一项所述的方法,其中溶液为含碱金属的溶液。
[0104]
e14.如e9
‑
e13中任一项所述的方法,其中诱导碳酸盐固体的沉淀包括诱导碳酸钙或碳酸镁中的至少一种的沉淀。
[0105]
虽然本公开已参考其特定实施方案进行了描述,但本领域的技术人员应当理解,在不脱离如一个或多个所附权利要求限定的本公开的真实精神和范围的情况下,可作出各种更改并且可替换等效物。此外,可进行许多修改以使特定情况、材料、物质组成、方法、一种或多种操作适于本公开的目的、精神和范围。所有这些修改都在一个或多个所附权利要求的范围内。具体地,尽管已参考以特定顺序执行的特定操作描述了某些方法,但是可以理解,在不脱离本公开的教导内容的情况下,这些操作可以被组合、细分或重新排序以形成等
效的方法。因此,除非本文明确说明,否则操作的顺序和分组不是对本公开的限制。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1