一种基于改性H-Beta分子筛的生物质氧化裂解催化剂及制备方法与应用
本发明涉及生物质化学转化利用领域,具体为一种基于改性h-beta分子筛的生物质氧化裂解催化剂及制备方法与应用。
背景技术:
生物质是地球上存在最广泛的物质,其主要成份——木质素和纤维素每年以约1640亿吨的速度再生,如以能量换算其相当于石油产量的15-20倍。同时,由于生物质是碳中性的,因此与化石资源相比,生物质是来源更为广泛的清洁可再生资源。
生物质的组成单体中大多都存在两个或多个相邻碳原子上含有羟基的结构,该结构类似1,2-二醇这样的结构单元。因此很多生物质的转化,都可从1,2-二醇结构单元的变化入手分析讨论。如果能在温和条件下实现对1,2-二醇的选择性氧化,断裂邻位二醇的c-c键生成对应的醛或酮,使生物质转化成小分子的高附加值产物,则将对生物质的利用产生深远的影响。
中国专利(申请号:201110032160.0)公开了一种使芳基1,2-二醇的c-c键断裂醇羟基转变为酮的氧化工艺。该方法使用液溴作为氧化剂,将2-甲基-1-苯基-1,2-丙二醇、α-(1-羟基环己基)-苯-甲醇、1-(4-甲氧基苯基)-2-甲基-1,2-丙二醇转化为相应的酮。所述氧化剂实现了芳基1,2-二醇的高转化率和高选择性。但由于液溴容易挥发,且有着极强烈的毒害性与腐蚀性,因此无法适应清洁生产的需求。同时由于液溴是均相氧化剂,因此其难以与反应体系分离,不易于回收和再利用。
公开文献coordinationchemistryreviews,2015,301-302:147-162.报道了一种使用氧气作为清洁氧化剂,由均相的钒基多金属氧酸盐作为催化剂实现了氢化苯偶姻、苯基乙二醇等1,2-二醇的氧化裂解,将1,2-二醇的c-c键断裂,醇羟基转变为相应的醛或者酯。该方法使用o2作为清洁的氧化剂,避免了有毒有害氧化剂的使用。公开文献journalofmolecularcatalysisa:chemical,1996,110(3):221-226.;journalofmolecularcatalysisa:chemical,2006,243(2):214-220.;organicletters,1999,1(5):713-715.;journalofmolecularcatalysisa:chemical,2000,156(1):279-281.;thejournaloforganicchemistry,2010,75(7):2321-2326.;advancedsynthesis&catalysis,2002,344(9):1017-1021.;catalysiscommunications,2008,9(6):1282-1285.;journaloftheamericanchemicalsociety,1988,110(4):1187-1196.等就使用o2作为氧化剂的均相催化体系进行了大量探索。然而这些均相催化剂不具备重复使用性,而且产物选择性较低。
因此,构建具有较高反应活性和选择性,同时反复使用性能优良的新的催化体系则成为一个需要攻克的难点。
技术实现要素:
为解决上述技术问题,本发明提供一种基于改性h-beta分子筛的生物质氧化裂解催化剂及制备方法与应用。
为实现上述目的,本发明采用如下的技术方案:
一种基于改性h-beta分子筛的生物质氧化裂解催化剂的制备方法,将活性组分前驱物溶解在去离子水中,得到活性组分溶液,然后将脱铝h-beta分子筛分散在活性组分溶液中进行搅拌浸渍,经旋转蒸发、干燥、焙烧后得到基于改性h-beta分子筛的生物质氧化裂解催化剂。
本发明进一步的改进在于,活性组分前驱物为铜盐、锌盐、铁盐、钴盐、银盐或金酸。
本发明进一步的改进在于,活性组分前驱物为硝酸铜、硫酸铜、氯化铜、硝酸锌、硫酸锌、氯化锌、硝酸铁、硝酸亚铁、硫酸铁、硫酸亚铁、氯化铁、氯化亚铁、硝酸银、氯化钴、硫酸钴、硝酸钴与四氯金酸中的一种或几种;
本发明进一步的改进在于,活性组分前驱物和脱铝h-beta分子筛的质量比为(0.01~1):1。
本发明进一步的改进在于,搅拌浸渍的速度为300~500r/min,搅拌浸渍的时间为1~48h;旋转蒸发的温度为40~80℃,压力为10~80kpa,转速为10~80r/min。
本发明进一步的改进在于,干燥温度为80~150℃,干燥时间为2~12h。
本发明进一步的改进在于,焙烧的具体条件为:以2℃/min的升温速率自室温升温至300~600℃,并保温4~6h。
一种根据上述方法制备的基于改性h-beta分子筛的生物质氧化裂解催化剂,该催化剂表面lewis酸酸量为200~800μmol(nh3)/g,
一种上述方法制备的基于改性h-beta分子筛的生物质氧化裂解催化剂的应用,将苯基乙二醇溶解在溶剂中,然后加入到高压反应釜,并加入催化剂,通过氧气加压至0.5~5mpa,在100~200℃下反应2~24h,得到氧化裂解产物。
本发明进一步的改进在于,溶剂是甲醇、乙醇、正丙醇、异丙醇、正丁醇与四氢呋喃中的一种或几种;苯基乙二醇与催化剂的质量比为1:(0.05~0.5)。
本发明进一步的改进在于,其中干燥温度为80~150℃,干燥时间为1~6h,焙烧的具体条件为:以5~20℃/min的升温速率自室温升温至300~600℃,并保温0.5~3h。
与现有技术相比,本发明具有的有益效果:
本发明选用的h-beta沸石是一种多孔的、结构规则且具有机械和化学稳定性的固体材料,可作为非均相反应的载体。h-beta沸石具有三维十二元环孔结构,该结构使h-beta沸石不仅拥有更大的比表面积和热稳定性,而且具有优异的离子交换性能,即脱铝后的h-beta沸石将允许金属前驱物填充到蚀刻铝留下的空位中。基于此,可以选择对邻位二醇催化活性更佳的金属组份与脱铝分子筛进行离子交换,并进入到脱铝分子筛的骨架中充当lewis酸和反应过程的吸附位点,进而利用金属离子的价态变化及其与h-beta载体的协同作用而调节其裂解活性与反应的选择性,从而构建出满足邻位二醇c-c键断裂的高效且可重复使用的催化剂,为具有1,2-二醇结构的生物质的利用,探索一条经济、高效、环保的新途径。本发明中采用脱铝沸石可以使活性组分可以进入蚀刻铝留下的空位中,提高催化剂的活性。
本发明以脱铝h-beta分子筛为载体,选择具有裂化活性的金属作为活性中心,采用浸渍法,通过旋蒸、干燥、焙烧等步骤,开发了一种基于改性h-beta分子筛的催化剂并用于生物质氧化裂解反应。该工艺中,通过不同活性组分前驱物的用量和制备方法,在保持催化剂载体织构性质基本不变的情况下,使活性组分进入到脱铝分子筛的骨架中。
本发明所得催化剂,一方面作为
该催化剂具有制备方法简单,原料价格低廉,工艺条件温和,反应过程绿色,催化剂可重复利用等优点,在生物质氧化裂解反应中有着独特的优势。
进一步的,采用2℃/min的升温速率较慢的升温速率,有利于脱铝分子筛载体的孔道结构控制,较快的升温速率会导致脱铝h-beta沸石载体的孔道结构改变。
进一步的,在300~600℃下保温4~6h的目的是使活性组分的可溶于水的前驱物,生成相应的氧化物,提高催化剂的活性。
本发明制备的催化剂用于生物质氧化裂解的反应,模型反应物苯基乙二醇的转化率较高,氧化裂解产物的选择性和收率较高,且催化剂具有可回收性。
附图说明
以下结合附图及具体实施方式对本发明作进一步的详细描述。
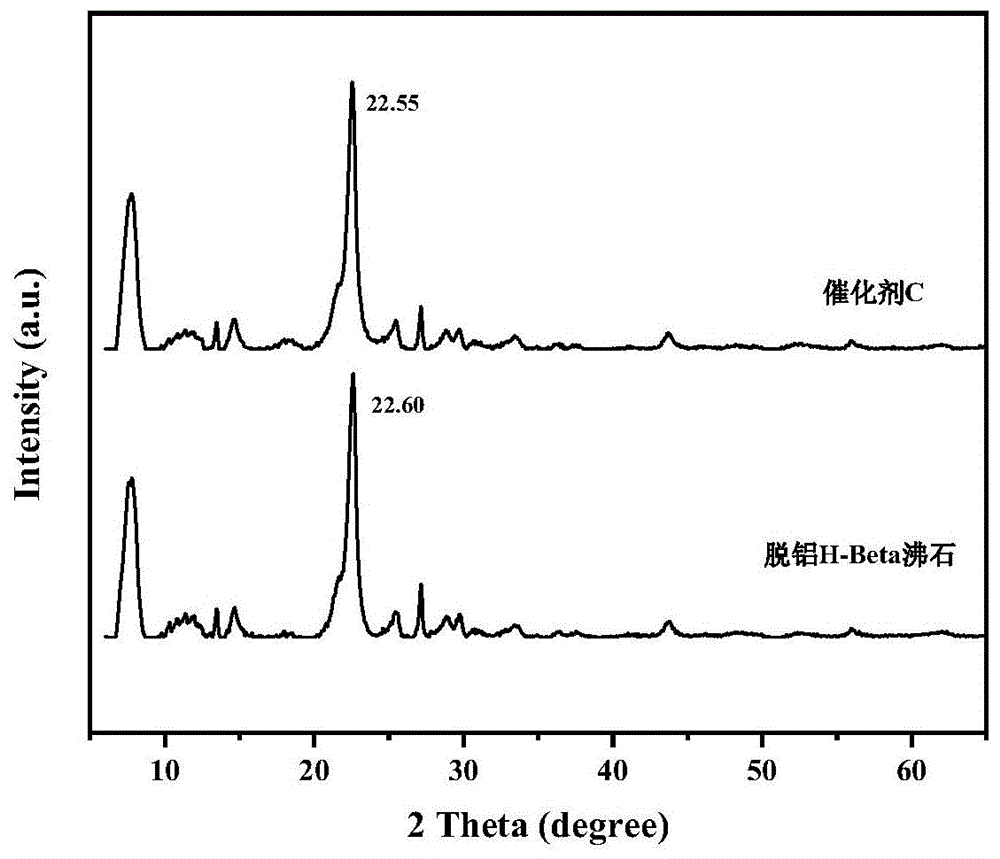
图1为实施例3制备的催化剂的x-射线衍射图。
图2为实施例3制备的催化剂的sem图。其中,(a)为低放大倍数,(b)为高放大倍数。
图3为实施例3制备的催化剂的nh3-tpd曲线。
图4为实施例3制备的催化剂的xps表征图。
图5为实施例7制备的催化剂的x-射线衍射图。
图6为实施例7制备的催化剂的sem图。其中,(a)为低放大倍数,(b)为高放大倍数。
图7为实施例16反应结束后的催化剂的sem图。其中,(a)为低放大倍数,(b)为高放大倍数。
图8为实施例16反应结束后的催化剂的xps表征图。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例,对本发明进一步详细说明。应当理解,此处所描述的具体实施例仅仅用于解释本发明,并不用于限定本发明。
本发明的一种基于改性h-beta分子筛的生物质氧化裂解催化剂的制备方法,包括以下步骤:
将可溶性活性组分前驱物溶解在去离子水中,得到活性组分溶液,然后将脱铝h-beta载体分散在活性组分溶液中进行搅拌浸渍,经旋转蒸发、干燥、焙烧后得到非均相的负载型催化剂。
其中,可溶性活性组分前驱物为硝酸铜、硫酸铜、氯化铜、硝酸锌、硫酸锌、氯化锌、硝酸铁、硝酸亚铁、硫酸铁、硫酸亚铁、氯化铁、氯化亚铁、硝酸银、氯化钴、硫酸钴、硝酸钴与四氯金酸中的一种或几种。
本发明进一步的改进在于,活性组分前驱物和脱铝h-beta载体的质量比为(0.01~1):1;搅拌浸渍的速度为300~500r/min,搅拌浸渍的时间为1~48h;旋转蒸发的温度为40~80℃,压力为10~80kpa,转速为10~80r/min。
本发明进一步的改进在于,干燥温度为80~150℃,干燥时间为2~12h,焙烧的具体条件为:以2℃/min的升温速率自室温升温至300~600℃,并保温4~6h。
以上所得生物质氧化裂解催化剂表面lewis酸酸量可达200~800μmol(nh3)/g,
一种基于改性h-beta分子筛的生物质氧化裂解催化剂在苯基乙二醇氧化裂解反应中的应用:
将苯基乙二醇溶解在一定量的溶剂中,然后将溶液和催化剂装入高压反应釜。将高压反应釜内的空气用氧气吹扫置换1~5次后,将氧气加压至0.5~5mpa。此后将反应釜转移至智能恒温定时磁力搅拌器中,待加热到所需的反应温度后,以500~1200r/min的速度进行搅拌开始反应。反应结束后得到氧化裂解产物。将高压反应釜浸入0~25℃的冷水中快速冷却,通过离心操作分离固体催化剂与氧化裂解的液体产物。分离的固体催化剂经过干燥、焙烧实现回收。
本发明进一步的改进在于,反应过程中的溶剂可以是甲醇、乙醇、正丙醇、异丙醇、正丁醇与四氢呋喃中的一种或几种。
本发明进一步的改进在于,苯基乙二醇与催化剂的质量比为1:(0.05~0.5)。
本发明进一步的改进在于,反应温度为100~200℃,反应时间为2~24h。
本发明进一步的改进在于,离心分离的转速为3000~10000r/min。
本发明进一步的改进在于,液体产物可以是苯甲醛、苯甲醇、苯甲酸、苯甲酸甲酯、苯甲酸乙酯与苯甲醛二甲缩醛中的一种或几种。
本发明进一步的改进在于,回收固体催化剂的干燥温度为80~150℃,干燥时间为1~6h,焙烧的具体条件为:以1~10℃/min的升温速率自室温升温至300~600℃,并保温0.5~3h。
下面为具体实施例。
实施例1
将活性组分前驱物溶解在30ml去离子水中,得到活性组分溶液,然后将4g脱铝h-beta载体分散在活性组分溶液中,并在400r/min的磁力搅拌下浸渍12h,后将得到的悬浊液在旋转蒸发仪上,在60℃的温度和-0.09mpa的压力下,以40r/min的转速旋转蒸发至干燥,再将得到的固体在烘箱中110℃干燥12h。干燥后,置于马弗炉中以2℃/min的升温速率从室温升温至450℃,焙烧6h,得到负载型催化剂,记为a。其中,活性组分前驱物为cu(no3)2·3h2o,活性组分前驱物和脱铝h-beta载体的质量比为5:100;
实施例2
与实施例1的不同之处在于,活性组分前驱物和脱铝h-beta载体的质量比为10:100;该实施例制备的催化剂记为b。
实施例3
与实施例1的不同之处在于,活性组分前驱物和脱铝h-beta载体的质量比为15:100;该实施例制备的催化剂记为c。
从图1中可以看出,实施例3制备得到的催化剂c,其x射线衍射图谱出峰位置为7.77°,11.39°,13.47°,14.70°,21.66°,22.55°,25.45°,27.14°,28.84°,29.72°,33.63°和43.68°,这与beta分子筛拓扑结构相对应,表明改性后催化剂载体织构性质基本不变。沸石骨架结构的膨胀,表现为衍射角2θ在22.55°附近的主衍射峰位置的变化。在改性后,主衍射峰的2θ值由22.60°减小至22.55°,表明beta沸石结构产生了扩张,金属已进入晶格。
从图2中(a)和(b)中可以看出,实施例3制备得到的催化剂为规则生长的粒径为500nm左右的球状结构。
从图3可以看出,实施例3制备得到的催化剂,其nh3-tpd脱附曲线中在不同温度下出现三个明显的脱附峰,脱附峰的温度对应不同表面酸性的强度,表明样品表面有不同强度的酸性中心。在100~250℃时的脱附带(弱吸附位点)可归因于弱吸附氨的解吸。在250~500℃时出现的脱附带(中等强度吸附位点)可归因于吸附在强lewis酸位点上的nh3。而550℃以上的脱附带(强吸附位点)可归因于吸附在
从图4可以看出,在cu2p光谱中,930–937ev和950–959ev处有到两个峰,分别对应于cu2p3/2和cu2p1/2。此外,在938–950ev和962–966ev范围内出现了两个携振卫星峰。由于cu(0),cu(ⅰ)的3d均为闭壳层,只出现一个终态;cu(ⅱ)的3d为开壳层,3d电子可以与o2p交叠耦合,产生终态分裂,使它出现两个终态,因此可以在主峰的高结合能处形成携振卫星峰。因此这表明实施例3制备的催化剂中存在cu(ⅱ)。
将cu2p3/2和cu2p1/2解卷积为两个峰。在952.5ev和932.7ev处的峰表示催化剂中存在cu-o-cu键结构。而在结合能较高的953.4ev和934.0ev处得到了与催化剂晶格氧原子配位的cu2+峰,即cu-o-si键结构。由于si比cu的电负性更强,cu-o-si中的cu-o键比cu-o-cu中的cu-o键具有更高的结合能。这些结果表明,cu2+和cuo都存在于实施例3制备的催化剂中,而且都可作为反应活性位点,从而可大幅增加催化剂c-c键的断裂能力。
实施例4
与实施例1的不同之处在于,活性组分前驱物和脱铝h-beta载体的质量比为20:100;该实施例制备的催化剂记为d。
实施例5
与实施例1的不同之处在于,活性组分前驱物和脱铝h-beta载体的质量比为25:100;该实施例制备的催化剂记为e。
实施例6
与实施例1的不同之处在于,活性组分前驱物和脱铝h-beta载体的质量比为50:100;该实施例制备的催化剂记为f。
实施例7
与实施例1的不同之处在于,活性组分前驱物和脱铝h-beta载体的质量比为100:100;该实施例制备的催化剂记为g。
从图5可以看出,实施例7制备得到的催化剂,其x射线衍射图谱出现了与beta分子筛拓扑结构相对应的衍射峰。同时,在35.52°,38.74°,48.77°,53.48°,58.31°和61.65°出现了对应于氧化铜晶型的衍射峰。
从图6中(a)和(b)可以看出,实施例7制备得到的催化剂为规则生长的粒径为500nm左右的球状结构。
实施例1-7制备的催化剂的比表面积、孔容积、活性金属的质量分数如表1所示。
表1催化剂样品的物理化学性质
a.bet比表面积和孔容积用氮气物理吸脱附测试测得;
b.活性金属的质量分数由初始配比经过计算得到。
由表1可以看出,实施例1-7制备的催化剂均具有较大的比表面积和孔容积,可以暴露更多的活性位点,有利于催化反应的发生。
实施例8
与实施例1的不同之处在于,活性组分前驱物和脱铝h-beta载体的质量比为1:100。
实施例9
与实施例3的不同之处在于,活性组分前驱物为cucl2。该实施例制备的催化剂记为h。
实施例10
与实施例3的不同之处在于,活性组分前驱物为fe(no3)3。该实施例制备的催化剂记为i。
实施例11
与实施例8的不同之处在于,活性组分前驱物为zn(no3)2。该实施例制备的催化剂记为g。
实施例12
与实施例8的不同之处在于,活性组分前驱物为cocl2。该实施例制备的催化剂记为k。
实施例13
与实施例8的不同之处在于,活性组分前驱物为四氯金酸。该实施例制备的催化剂记为l。
实施例14
同实施例1,与实施例1不同在于,活性组分前驱物为硝酸铜与硫酸铜的混合物。
实施例15
同实施例1,与实施例1不同在于,活性组分前驱物为硝酸铜与氯化铜的混合物。
载型催化剂在苯基乙二醇氧化裂解反应中的应用:
实施例16
将138mg苯基乙二醇溶解在2ml的甲醇中,得到溶液,然后将溶液和实施例3中的催化剂50mg装入高压反应釜。将高压反应釜内的空气用氧气吹扫置换3次后,将氧气加压至3.0mpa。此后将反应釜转移至智能恒温定时磁力搅拌器中,控制反应温度为150℃,并在800r/min下的速度进行搅拌,反应8h反应结束后得到氧化裂解产物。
将高压反应釜浸入4℃的冷水中快速冷却,后将得到的悬浊液通过离心操作,在8000r/min的转速下分离出固体催化剂,得到苯甲醛、苯甲醇、苯甲酸甲酯、苯甲醛二甲缩醛四种液相产物。将分离的固体催化剂在120℃干燥2h,然后以10℃/min的升温速率自室温升温至450℃,并保温1h,实现催化剂的回收。
从图7中(a)和(b)可以看出,反应后的催化剂为粒径500nm左右的球状颗粒,其结构和形貌在反应前后基本保持稳定。
从图8可以看出,使用后的催化剂在主峰的高结合能处仅得到了两个较弱的携振卫星峰,这说明在反应过程中,部分cu由+2价变为了+1价。这个结果证实了在反应过程中吸附在cu上的具有亲核性的o2-物种可以插入1,2-二醇中缺电子的位置上,在反应物c-c键断裂的同时,cu-o键发生断裂,cu可以得到电子被还原为+1价。
实施例17
与实施例16的不同在于,将溶液和实施例1中的催化剂50mg装入高压反应釜。
实施例18
与实施例16的不同在于,将溶液和实施例2中的催化剂50mg装入高压反应釜。
实施例19
与实施例16的不同在于,将溶液和实施例4中的催化剂50mg装入高压反应釜。
实施例20
与实施例16的不同在于,将溶液和实施例5中的催化剂50mg装入高压反应釜。
实施例21
与实施例16的不同在于,将溶液和实施例6中的催化剂50mg装入高压反应釜。
实施例22
与实施例16的不同在于,将溶液和实施例7中的催化剂50mg装入高压反应釜。
实施例17-22中各产物的收率和氧化裂解产物总收率如表2所示。
表2催化剂性能评价
实施例23
与实施例16的不同之处在于,使用实施例9中的催化剂。所得产物为苯甲醛、苯甲醇、苯甲酸甲酯、苯甲醛二甲缩醛。
实施例24
与实施例16的不同之处在于,使用实施例10中的催化剂。所得产物为苯甲醛、苯甲醇、苯甲酸、苯甲酸甲酯、2-甲氧基-2-苯基-乙酸乙酯。
实施例25
与实施例16的不同之处在于,使用实施例11中的催化剂,且控制反应温度为120℃。所得产物为苯甲醛、苯甲醛二甲缩醛。
实施例26
与实施例16的不同之处在于,使用实施例12中的催化剂,且控制反应温度为120℃。所得产物为苯甲醛二甲缩醛、苯乙酸甲酯。
实施例27
与实施例16的不同之处在于,使用实施例13中的催化剂,且控制反应温度为120℃。所得产物为苯甲醛二甲缩醛。
实施例23-27中各产物的收率和氧化裂解产物总收率如表3所示。
表3催化剂性能评价
实施例28
与实施例16的不同在于,催化剂用量为10mg。
实施例29
与实施例16的不同在于,催化剂用量为20mg。
实施例30
与实施例16的不同在于,催化剂用量为30mg。
实施例31
与实施例16的不同在于,催化剂用量为40mg。
实施例28-31中各产物的收率和氧化裂解产物总收率如表4所示。
表4催化剂性能评价
实施例32
与实施例16的不同在于,将溶液和实施例1中的催化剂6.9mg装入高压反应釜。
实施例33
与实施例16的不同在于,将溶液和实施例1中的催化剂69mg装入高压反应釜。
实施例34
与实施例16的不同在于,将氧气加压至0.5mpa。
实施例35
与实施例16的不同在于,将氧气加压至1.0mpa。
实施例36
与实施例16的不同在于,将氧气加压至2.0mpa。
实施例34-36中各产物的收率和氧化裂解产物总收率如表5所示。
表5催化剂性能评价
实施例37
与实施例16的不同在于,将氧气加压至4mpa。
实施例38
与实施例16的不同在于,将氧气加压至5mpa。
实施例39
与实施例16的不同在于,控制反应温度为120℃。
实施例40
与实施例16的不同在于,控制反应温度为130℃。
实施例41
与实施例16的不同在于,控制反应温度为140℃。
实施例39-41中各产物的收率和氧化裂解产物总收率如表6所示。
表6催化剂性能评价
实施例42
与实施例16的不同在于,控制反应温度为100℃,反应时间为24h。
实施例43
与实施例16的不同在于,控制反应温度为200℃,反应时间为2h。
实施例44
与实施例16的不同在于,控制反应温度为170℃,反应时间为10h。
实施例45
与实施例16的不同在于,控制反应温度为160℃,反应时间为20h。
实施例46
与实施例16的不同在于,将138mg苯基乙二醇溶解在2ml的乙醇中。所得产物为苯甲醛、苯甲醇、苯甲酸、苯甲酸乙酯、2-甲基-4-苯基-1,3-二氧戊环。
实施例47
与实施例16的不同在于,将138mg苯基乙二醇溶解在2ml的正丙醇中。所得产物为苯甲醛、苯甲醇、苯甲酸、苯甲酸丙酯。
实施例48
与实施例16的不同在于,将138mg苯基乙二醇溶解在2ml的异丙醇中。所得产物为苯甲醛、苯甲醇、苯甲酸。
实施例49
与实施例16的不同在于,将138mg苯基乙二醇溶解在2ml的正丁醇中。所得产物为苯甲醛、苯甲醇、苯甲酸。
实施例50
与实施例16的不同在于,将138mg苯基乙二醇溶解在2ml的四氢呋喃中。所得产物为苯甲酸。
实施例16、46-50中各产物的收率和氧化裂解产物总收率如表7所示。
表7催化剂性能评价
本发明中将活性组分引入脱铝h-beta的骨架中作为活性位点,利用改性材料中
以上所述,以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施例对本发明进行了详细地说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的精神和范围。
- 还没有人留言评论。精彩留言会获得点赞!