一种双功能化石墨烯负载NiAuPd纳米催化剂的制备方法及应用
一种双功能化石墨烯负载niaupd纳米催化剂的制备方法及应用
技术领域
1.本发明涉及催化剂制备以及环境和能源的可持续发展领域,特别是一种双功能化石墨烯负载niaupd纳米催化剂的制备方法及应用。
背景技术:
2.当今世界能源需求主要依靠化石燃料,但是化石燃料具有不可再生,污染环境严重等问题。因此开发新型可持续能源迫在眉睫。氢能被认为是有效的满足人类能源需求的清洁能源,尤其是与燃料电池技术的结合,可以为汽车、移动设备提供高效清洁的能源动力,具有重要的意义。
3.目前,储氢方式一般可分为物理与化学两种方法。除了以压缩气体、低温液体的形式存储h2或使用氢存储材料(例如金属氢化物)之外,化学存储材料(水合肼n2h4和甲酸hcooh等)由于氢含量高且易于以液体形式充电,因此成为主要关注点。其中甲酸被认为是化学储氢的一种有希望的替代品,因为它的液体材料对人体和环境无毒,并且具有高氢含量(4.4wt%),易于储存和运输,被认为是生产和储存氢气的潜在载体。甲酸在适当催化剂作用下可以发生脱氢反应,生成需要的氢气和二氧化碳(hcooh
→
h2+co2);然而甲酸也有可能发生不被期望的脱水反应,生成水和一氧化碳(hcooh
→
h2o+co),co对燃料电池催化剂有毒,是一种不希望发生的副反应,必须严格控制。对于甲酸脱氢反应的产物,除了h2之外,所产生的co2可以再通过光催化或者电催化的方式再生成甲酸,这就实现了一个碳中性循环。
4.与均相催化剂相比,异相催化剂由于其具有容易控制、方便提取、便于回收等优点而被广泛地研究与使用。pd纳米颗粒对于催化甲酸脱氢反应具有非常好的效果,但是由于其资源稀缺和价格昂贵,并不适合大规模应用。通过与成本低廉的ni、co等过渡族金属nps进行复合,所形成的合金或者核壳结构的nps,不近降低了催化剂的成本,而且还能够提高催化性能,因此是一种有效的改善催化性能的方式。此外,合适的载体材料对于催化性能的改善也具有非常明显的效果,因为它能够有效的减小颗粒尺寸,降低团聚及改善金属颗粒在其上的分布情况。石墨烯(rgo)具有比表面积大,稳定性好,电子迁移能力强等优点,是一种良好的载体材料。但是石墨烯自身的亲水性较差,通过对石墨烯进行异质原子(b、n、p等)掺杂或功能化,能够有效的改善其亲水性和对金属离子的分散性。但是对于氮或者氨基的引入通常需要添加氨水等有毒的化学物质或者使用高温水热法等,这些方法只能实现单元素的掺杂且制备方法复杂。
5.综上所述,寻找一种简单且有效的方法合成价格低廉、高效且分散性好的纳米催化剂对于降低催化剂成本并提高甲酸脱氢反应效率是非常必要的。
技术实现要素:
6.本发明的目的是要解决现有技术中存在的不足,提供一种双功能化石墨烯负载niaupd纳米催化剂的制备方法及应用。与常用的水热法相比,该方法采用简单的一步共还
原法,在室温下快速的制备出了氮磷双功能化的石墨烯负载的金属纳米催化剂。本发明在催化甲酸室温水解制氢反应中,2分钟内可实现甲酸的完全分解,具有极高的催化活性,且转换率为100%,氢气选择性为100%。
7.为达到上述目的,本发明是按照以下技术方案实施的:
8.一种双功能化石墨烯负载niaupd纳米催化剂的制备方法,该方法包括如下步骤:
9.步骤一、采用hummer’s方法预先制备出氧化石墨烯go,将制得的go加水稀释,配制浓度1~10mg/ml的go水溶液,超声均匀;
10.步骤二、将3
‑
氨丙基三乙氧基硅烷apts和磷酸氢二钾k2hpo4加入到go水溶液中,搅拌10~20min得到溶液a;
11.其中,每10~50mg go中加0.1~0.8ml的3
‑
氨丙基三乙氧基硅烷apts和50~150mg的磷酸氢二钾k2hpo4;
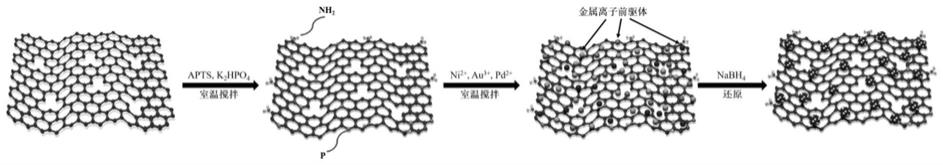
12.步骤三、将nicl2、haucl4和na2pdcl4水溶液依次添加到步骤一所述的溶液a中,继续搅拌1~10min后,得到混合溶液b;
13.其中,摩尔比为,nicl2:haucl4:na2pdcl4=1:1~3:1~8(优选为1:1:3);所述的的浓度为0.01~1m;每15~25ml溶液b中含0.02~0.1mmol的nicl2;
14.步骤四、将硼氢化钠nabh4作为还原剂加入到步骤三所述的混合溶液b中,搅拌还原,得到混合溶液c;
15.每15~25ml溶液b加入15~150mg硼氢化钠nabh4;
16.步骤五、在室温下,将步骤四所述的混合溶液c在空气中磁力搅拌还原,待没有气泡时,7000~12000rpm离心4~10min,水洗,即可得到所需的双功能化石墨烯负载niaupd纳米催化剂(niaupd/bi
‑
fg)。
17.前述步骤五所述的niaupd/bi
‑
fg催化剂中,niaupd为三金属合金结构,且均匀的分散在双功能化石墨烯载体上,颗粒尺寸为1.05~2.25nm。
18.前述步骤三中的nicl2溶液、haucl4溶液、na2pdcl4水溶液的浓度为0.01~1m。
19.前述步骤四中的nabh4进行还原反应的温度为室温。
20.前述的一种双功能化石墨烯负载niaupd纳米催化剂,用于催化甲酸室温水解制氢反应。
21.具体包括如下步骤:包括如下步骤:将得到的催化剂分散在水中,再加入甲酸水溶液,温度25~50℃、常压,即可催化甲酸水解制氢;
22.其中,每5~15ml水中加入0.1~1mmol催化剂;甲酸水溶液的浓度为0.1~5m,催化剂和甲酸的摩尔比为0.01~0.5:1;催化剂的摩尔量以ni、au和pd三种元素的摩尔量之和计。
23.与现有技术相比,本发明的有益效果是:
24.本发明的一种双功能化石墨烯负载niaupd纳米催化剂的方法,采用一步共还原法快速的制备双功能化石墨烯负载niaupd纳米催化剂,在室温下即可完成,具有合成时间短、操作简便等优点,且明显提高了niaupd nps在bi
‑
fg衬底上的分散性及降低了金属nps的颗粒尺寸;将合成的niaupd/bi
‑
fg催化剂用于催化甲酸水溶液室温分解制氢,该催化剂在没有任何添加剂存在的条件下仍然具有极高的催化活性,100%的转化率,100%的氢气选择性及较好的循环稳定性和极高的催化活性,在2分钟内可以实现甲酸的完全分解,其tof值
高达4554.75h
‑1,其性能远高于目前已经报道的大部分nipd/nh2‑
n
‑
rgo(转换率100%,tof为954.3h
‑1)、pd/c(转换率95%,tof为913h
‑1)、niaupd/f
‑
gns(转换率100%,tof为1090h
‑1)等催化剂,且niaupd/bi
‑
fg中添加了非贵金属,有效的降低了催化剂的成本。总的来说,该方法能够作为一种新的简单的功能化的方法,来合成具有高效催化性能的双功能化石墨烯负载niaupd纳米催化剂,将所合成的催化剂应用于甲酸在室温下分解制氢反应,为发展安全、高效、廉价的催化剂提供新的途径,并进一步促进甲酸作为储氢材料在实际生活中的应用。
附图说明
25.图1为实施例1中niaupd/bi
‑
fg催化剂的制备示意图;
26.图2为实施例1中niaupd/bi
‑
fg催化剂的x射线衍射图谱;
27.图3为实施例1中niaupd/bi
‑
fg催化剂的x射线光电子能谱;其中,图3a为ni 2p的x射线光电子能谱,图3b为au 4f的x射线光电子能谱,图3c为pd 3d的x射线光电子能谱,图3d为c1s的x射线光电子能谱,图3e为n1s的x射线光电子能谱,图3f为p 2p的x射线光电子能谱;
28.图4为实施例1中niaupd/bi
‑
fg、比较例1中niaupd/g催化剂透射电镜图片;其中,图4a为niaupd/bi
‑
fg的透射图片,图4b为niaupd/g的透射图片;
29.图5为实施例1中niaupd/bi
‑
fg、比较例1中niaupd/g、比较例2中niaupd/ng、比较例3中niaupd/pg催化剂在室温下催化甲酸分解的时间
‑
过程曲线;
30.图6为niaupd/bi
‑
fg催化甲酸分解的循环性能曲线图。
具体实施方式
31.下面结合具体实施例对本发明作进一步描述,在此发明的示意性实施例以及说明用来解释本发明,但并不作为对本发明的限定。
32.实施例1
33.1.如图1所示,一种双功能化石墨烯负载niaupd基纳米催化剂的制备方法具体包括以下步骤:
34.将50mg go超声分散到14.6ml水中,超声后混合均匀,再加入0.4ml的3
‑
氨丙基三乙氧基硅烷apts和100mg的磷酸氢二钾k2hpo4,搅拌15min使之均匀。再将0.02mmol、0.02mmol和0.06mmol的nicl2、haucl4和na2pdcl4溶液(浓度均0.02m,体积分别为1ml,1ml,3ml)依次加入到上述混合溶液中(混合后总体积为20ml),继续搅拌5min使之均匀;40mg的nabh4在室温下加入到上述混合溶液中,磁力搅拌均匀,待没有气泡时,离心(8000rpm,离心5min)、水洗3次,制得niaupd/bi
‑
fg催化剂。
35.2.样品检测
36.(1)将前述制得的niaupd/bi
‑
fg催化剂真空干燥,研磨成细小粉末;参考图2,x射线粉末衍射(xrd)结果表明,该实验方法成功的合成了双功能化碳石墨烯负载的niaupd金属催化剂,且niaupd以合金的形式存在。
37.(2)将前述制得的niaupd/bi
‑
fg催化剂真空干燥;参考图3,x射线光电子能谱(xps)结果显示,该方法成功的合成了niaupd/bi
‑
fg,且ni,au,pd均主要以金属态存在,其
中ni有明显的氧化峰,可能是由于样品制备过程中的氧化造成的。
38.(3)将前述制得的niaupd/bi
‑
fg催化剂稀释,滴在碳支持膜上,干燥;参考图4a中透射电子显微镜(tem)结果显示,niaupd/bi
‑
fg样品具有较小的颗粒尺寸(~1.65nm)和均匀的分散性。
39.3.催化甲酸脱氢反应
40.将前述全部制得的niaupd/bi
‑
fg催化剂(催化剂的摩尔量以ni、au和pd三种元素的摩尔量之和计,即催化剂的摩尔数0.1mmol)分散到10ml水中,再加入5mmol(1m)的甲酸水溶液,并通过气体量管测量所产生的氢气。此次niaupd/bi
‑
fg催化剂催化甲酸水溶液制氢过程的制氢量(ml)与时间(分钟)图如图5中的(a)所示,催化甲酸室温水解制氢能够在2分钟内产生气体的量为245ml,转换率达到100%。
41.在第一轮分解反应结束以后,再将等量的甲酸溶液加入到双颈烧瓶当中,剩下的操作与之前反应的相同。将同样的操作步骤在25℃水浴温度下再重复五次,如图6所示,所制备的niaupd/bi
‑
fg纳米催化剂对于催化甲酸脱氢反应具有良好的循环稳定性。
42.比较例1(不加入apts和k2hpo4)
43.将50mg go分散到15ml水中,超声后混合均匀,再依次加入0.02mmol、0.02mmol和0.06mmol的nicl2、haucl4和na2pdcl4溶液(0.02m),混合后总体积为20ml,搅拌5min使之均匀;40mg的nabh4在室温下加入到上述混合溶液中,磁力搅拌均匀,待没有气泡时,还原完全后,离心(8000rpm,离心5min)、水洗3次,制得niaupd/g催化剂。
44.将niaupd/g催化剂分散到10ml水中,再加入5mmol(1m)的甲酸水溶液,并通过气体量管测量所产生的氢气。此次niaupd/g催化剂催化甲酸制氢过程的制氢量(ml)与时间(分钟)图如图5中的(b)所示,niaupd/g对于甲酸分解没有催化活性。
45.图4为实施例1和比较例1制得的样品的tem照片。从图中可以看出,在niaupd/bi
‑
fg中niaupd纳米颗粒都较均匀的分布在载体上,且颗粒较小;而在niaupd/g中,纳米颗粒在石墨烯载体上出现了明显的团聚,且颗粒较大。
46.比较例2(不加k2hpo4)
47.将50mg go分散到14.6ml水中,超声后混合均匀,加入0.4ml的3
‑
氨丙基三乙氧基硅烷apts,搅拌15min使之均匀。再将0.02mmol、0.02mmol和0.06mmol的nicl2、haucl4和na2pdcl4溶液(0.02m)依次加入到上述混合溶液中(混合后总体积为20ml),继续搅拌5min使之均匀;40mg的nabh4在室温下加入到上述混合溶液中,磁力搅拌均匀,待没有气泡时,还原完全后,离心(8000rpm,离心5min),离心、水洗3次,制得niaupd/ng催化剂。
48.将niaupd/ng催化剂分散到10ml水中,再加入5mmol(1m)的甲酸水溶液,并通过气体量管测量所产生的氢气。此次niaupd/ng催化剂催化甲酸水溶液制氢过程的制氢量(ml)与时间(分钟)图如图5中的(c)所示,催化甲酸室温水解制氢能够在3分钟内产生气体的量为245ml,转换率达到100%。
49.比较例3(不加入apts)
50.将50mg go分散到15ml水中,加入100mg的磷酸氢二钾k2hpo4,搅拌15min使之均匀。再将0.02mmol、0.02mmol和0.06mmol的nicl2、haucl4和na2pdcl4溶液(0.02m)依次加入到上述混合溶液中(混合后总体积为20ml),继续搅拌5min使之均匀;40mg的nabh4在室温下加入到上述混合溶液中,磁力搅拌均匀,待没有气泡时,还原完全后,离心(8000rpm,离心5min)、
水洗3次,制得niaupd/pg催化剂。
51.将niaupd/pg催化剂分散到10ml水中,再加入5mmol(1m)的甲酸水溶液,并通过气体量管测量所产生的氢气。此次niaupd/pg催化剂催化甲酸水溶液制氢过程的制氢量(ml)与时间(分钟)图如图5中的(d)所示,催化甲酸室温水解制氢的转化率为4.1%。
52.实施例2
53.其他步骤同实施例1,不同之处为将ni:au:pd的摩尔量改为1:1:8;
54.得到的产品的结构仍为niaupd三金属合金,性能与实施例1接近。
55.实施例3
56.其他步骤同实施例1,不同之处为将ni:au:pd的摩尔量改3:3:4;
57.得到的产品的结构仍为niaupd三金属合金,性能与实施例1接近。
58.总的来说,该方法能够作为一种新型、简单的制备双功能化石墨烯负载金属催化剂的方法,将所合成的催化剂应用于甲酸分解制氢反应之中,表现出优异的催化效果,为发展高效、稳定、低成本的金属纳米催化剂提供新的途径,并且促进了fa作为储氢材料在燃料电池、电动汽车、可穿戴设备等领域的实际应用。
59.本发明的技术方案不限于上述具体实施例的限制,凡是根据本发明的技术方案做出的技术变形,均落入本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1