β-环糊精金属有机骨架材料HPLC柱的制备及应用
β
‑
环糊精金属有机骨架材料hplc柱的制备及应用
技术领域
1.本发明涉及手性高效液相色谱(hplc)柱材料的制备及应用技术领域,具体涉及二种β
‑
环糊精(β
‑
cd)金属有机骨架(metal
‑
organic framework(mofs))材料hplc手性柱材料的制备及应用,即将β
‑
cd
‑
mofs材料制备成hplc手性柱的方法及对手性农药和手性药物对映异构体的拆分研究和应用。
背景技术:
2.日益增长的材料科学需求促使各种新材料不断被发现,金属有机框架(metal
‑
organic frameworks,mofs)材料凭借着灵活的可塑性和优异的功能性取得前所未有的发展。mofs优异的功能性主要体现在大的比表面积、高的孔隙率以及结构的多样性。可塑性主要表现为可根据应用需求改变合成条件获得相应的组成和结构。
3.近十年,对于mofs及其mofs复合材料的研究一直处于上升状态。迄今为止,mofs已有成千上万种不同的成分,在吸附与分离、催化、传感、载药等领域均有涉及。
4.具有手性空间群的mofs称为手性mofs。1999年aoyama等构筑出手性mofs,此后手性mofs优异的性能逐渐被人们了解。丰富可修饰的基团、错综复杂的手性环境使手性mofs在手性领域的应用十分活跃。为充分挖掘和发挥手性mofs的作用,研发合成新型手性mofs变得异常重要。
5.手性mofs的合成一般有直接合成、手性模板剂合成以及后合成三种。直接合成法的合成思路有两种,一是基于纯手性配体的直接合成,二是自发拆分。纯手性配体直接合成是使用如环糊精、氨基酸、手性冠醚等作为手性配体与金属离子直接形成手性mofs。相比纯手性配体直接合成,自发拆分形成的手性mofs具有更大的不可预测性,在设计和合成中有很大的挑战。手性模板剂合成是在合成mofs时额外加入手性模板剂来诱导无手性环境的mofs生成手性mofs,这种方法对于有序结构的合成更具优势。后合成是先合成带有
‑
cooh、
‑
ch2、
‑
nh2等取代基的非手性mofs,再加入手性试剂,通过共价键、氢键、静电相互作用等连接形成手性mofs。
6.在hplc系统中分离对映异构体需要流动相或固定相中包含有手性选择剂。显然,以固定的形式使用手性选择剂具有更大的意义。手性固定相(chiral stationary phase,csp)优选保留一种对映异构体的过程是将对映异构体转化为瞬态非对映异构配合物,常用的解释机制是“三点作用”,即对映体能被拆分的前提是对映体至少与csp产生三种以上的作用力。hplc
‑
csp被认为是拆分外消旋化合物的最佳方法之一。作为手性hplc的核心,csp至今为止已经研发出了很多种类型。根据手性选择剂的类型,可分为天然csp、半合成csp和合成csp三种。随着手性mofs种类不断增加,以手性mofs作为csp拆分对映异构体也得到广泛研究。自2007年,基于l
‑
乳酸手性配体合成mofs[zn2(bdc)(l
‑
lac)(dmf)]﹒dmf材料作为hplc
‑
csp拆分手性亚砜的报道之后,出现了各种类型的手性mofs材料用于hplc
‑
csp。例如氨基酸手性mofs、环糊精手性mofs、二氧化硅复合手性mofs和其他手性mofs。
[0007]
基于cd的mofs及其衍生物已被证明在对映体识别上有良好的发展前景。当前关于
cd
‑
mofs制备hplc
‑
csp的报道较少。2010年,stoddart报道了基于γ
‑
cd和碱金属阳离子合成的cd
‑
mofs。与其他手性mofs相比,cd
‑
mofs具有较大的表面积,丰富的手性识别位点(以γ
‑
cd为例,手性识别位点源自每个γ
‑
cd环中存在的40个立体异构中心)以及可利用的游离羟基通过氢键与手性分子相互作用,增强其对映体选择性识别的能力。2016年,hartlieb等使用γ
‑
cd
‑
mofs材料直接填装制备hplc
‑
csp柱,成功拆分了蒎烯对映异构体。2017年yang等也利用γ
‑
cd
‑
mofs材料直接填装制备hplc
‑
csp柱,以包括(r,s)
‑
α
‑
环丙基苄醇,(r,s)
‑1‑
苯基乙醇,(r,s)
‑
α
‑
乙烯基苄醇等十二种手性芳香醇为拆分对象,研究γ
‑
cd
‑
mofs的手性识别能力。结果显示,这十二种供试手性芳香醇有良好的拆分效果。zhen等以铜盐为金属离子,辛基
‑
[6
‑
脱氧
‑6‑
(3
‑
巯基丙酸钠)]
‑
γ
‑
环糊精为有机配体,以聚多巴胺为桥连支撑合成cu
‑
cd
‑
mofs,制备成毛细管电色谱(cec)csp。该csp对dl型的苯丙氨酸,亮氨酸,缬氨酸,苏氨酸,丝氨酸表现出理想的手性识别能力。
[0008]
基于cd
‑
mofs制备hplc
‑
csp的研究报道仍有限,值得本领域技术人员继续深入研究。
技术实现要素:
[0009]
针对现有技术中存在的不足,本发明的目的在于提供了一种新型手性高效液相色谱(hplc)柱材料的制备及应用技术,即将β
‑
cd
‑
mofs材料制备成高效液相色谱(hplc)手性柱的方法及对手性农药和手性药物对映异构体的拆分研究。
[0010]
本申请人将二种β
‑
cd
‑
mofs材料用于高效液相色谱领域,与硅胶基质键合后,作为手性柱材料可用于9种手性对映异构体的拆分。首次选用掺杂官能团的办法来合成β
‑
cd
‑
mofs材料。即将β
‑
环糊精(β
‑
cd)和间羧基苯磺酰氯作为有机配体,钾离子为中心金属离子合成手性mofs材料。首次通过“一锅法”和蒸汽扩散法分别构筑了具有不同空隙结构的手性mofs,并键合硅胶为hplc固定相填料,制备成hplc手性柱。该色谱柱能成功应用于手性农药和手性药物的对映体分离领域,结果表明,β
‑
cd
‑
mofs材料hplc柱对这些化合物均表现出较高的对映体选择性。该方法具有合成条件温和、绿色环保、快速、简单、成本低等优点。此项工作为9种手性化合物建立了新的hplc拆分体系,同时也证明了β
‑
cd
‑
mofs材料作为一种新的手性选择剂在手性分离分析方面具有的巨大潜力。
[0011]
具体涉及了二种β
‑
cd
‑
mofs材料。这些工作是基于以下步骤进行:(1)、β
‑
环糊精(β
‑
cd)和间羧基苯磺酰氯为有机配体,钾离子为中心金属离子作为合成手性mofs的原材料。通过“一锅法”和蒸汽扩散法构筑具有不同空隙结构的手性mofs,得到二种不同结构的手性mofs材料,分别命名为β
‑
cd
‑
mofs
‑
p50和β
‑
cd
‑
mofs
‑
p20。
[0012]“一锅法”的具体做法是:
[0013]
将β
‑
cd、间羧基苯磺酰氯和氯化钾溶于甲醇水溶液中,加入ctab调控颗粒度,密封静置24小时以上,即可获得β
‑
cd
‑
mofs
‑
p50材料;
[0014]
β
‑
cd、间羧基苯磺酰氯、氯化钾质量比为(0.1
‑
1.0):(0.1
‑
1.0):(0.1
‑
0.5),优选质量比为(0.8
‑
0.9):0.5:(0.2
‑
0.4),最佳质量比为0.7:0.5:0.3;
[0015]
ctab的加入量为β
‑
cd、间羧基苯磺酰氯和氯化钾总质量的6
‑
10%,最佳为8%。
[0016]
蒸汽扩散法的具体做法是:
[0017]
将β
‑
cd、间羧基苯磺酰氯和氯化钾溶在甲醇水溶液中,加入ctab调控颗粒度,过滤
不溶物后置于大的密封容器中,水浴(45
‑
70℃均可)加热反应至少3h,停止反应冷却后静置即可得到无色立方晶体,得晶体即为β
‑
cd
‑
mofs
‑
p20材料;
[0018]
β
‑
cd、间羧基苯磺酰氯、氯化钾质量比为(0.01
‑
0.8):(0.01
‑
0.8):(0.01
‑
0.5),优选质量比为0.1:(0.06
‑
0.09):(0.04
‑
0.06),最佳质量比为0.1:0.078:0.046。
[0019]
ctab的加入量为β
‑
cd、间羧基苯磺酰氯和氯化钾总质量的50
‑
60%,最佳为54%。
[0020]
通过固体紫外漫反射光谱、傅里叶红外光谱、x射线粉末衍射、核磁共振氢谱、x
‑
射线光电子能谱以及场发射电镜和光学显微镜等手段确定β
‑
cd
‑
mofs
‑
p50和β
‑
cd
‑
mofs
‑
p20的结构和形貌。以β
‑
cd
‑
mofs
‑
p50和β
‑
cd
‑
mofs
‑
p20键合硅胶为固定相填料,通过x
‑
射线光电子能谱和场发射电镜对柱1和柱2的固定相填料进行表征,证实了基于目标mofs的固定相材料已制备得到。用匀浆湿法填充制备hplc手性柱1和柱2。(2)、通过适当的表征手段寻找手性填料制备的最佳配方及反应。(3)用β
‑
cd
‑
mofs
‑
p20所制得的手性柱2可建立噁唑禾草灵和马来酸氯苯那敏二种物质的单一对映体的制备方法。(4)用所制得的二种β
‑
cd
‑
mofs手性柱可拆分手性药物和手性农药,建立了噁唑禾草灵、氟雷拉纳、喹禾灵、联苯菊酯、雷诺嗪、奥硝唑、多沙唑嗪、羟丙哌嗪和马来酸氯苯那敏等手性化合物的对映体拆分和定量分析方法,部分手性药物可通过该手性柱制备获得单一对映体。
[0021]
本发明利用β
‑
cd
‑
mofs材料制成手性固定相填料,可建立多种手性物质单一对映体的hplc拆分及定性定量检测新方法。结果显示此种制备hplc柱的方法是有效的,可成功地将β
‑
cd
‑
mofs键合到硅胶基质上,再填充至hplc柱内,也进一步表明此种hplc柱制备法是可行的。用上述β
‑
环糊精金属有机骨架材料制备手性hplc柱的方法是有创新性的工作,在hplc手性固定相中运用的相关工作也是有创新性的。
[0022]
与现有技术相比,本发明的优点和有益效果在于:
[0023]
1、首次用β
‑
环糊精金属有机骨架材料作为hplc固定相对手性化合物进行拆分,未见文献报道,该技术弥补了这一领域的空白。
[0024]
2、提供了一种新型β
‑
环糊精掺杂有机官能团的新型mofs材料的合成方法,由此可制备高效液相色谱柱填料,确定了用该填料,制备手性hplc柱的方法,拓展了这一物质的应用范围。
[0025]
3、用β
‑
cd
‑
mofs材料制成的制备型手性hplc柱从消旋体中可得到纯化的噁唑禾草灵和马来酸氯苯那敏单一对映体纯品,为这两种手性物质的单一对映体制备提供了一种新方法。
[0026]
4、在β
‑
cd
‑
mofs手性hplc柱上获得了可分离噁唑禾草灵、氟雷拉纳、喹禾灵、联苯菊酯、雷诺嗪、奥硝唑、多沙唑嗪、马来酸氯苯那敏和羟丙哌嗪等手性物质的分离条件,在最佳条件下可实现基线分离,据此可建立多种手性物质单一对映体hplc定量测定新方法。
附图说明
[0027]
图1:β
‑
cd
‑
mofs
‑
p50材料的扫描电镜图;
[0028]
图2:β
‑
cd
‑
mofs
‑
p20材料的扫描电镜图;
[0029]
图3:β
‑
cd
‑
mofs材料的x射线粉末衍射(xrd)图;其中,(a)
‑
间羧基苯磺酰氯;(b)
‑
β
‑
cd;(c)
‑
β
‑
cd
‑
mofs
‑
p50材料;(d)
‑
β
‑
cd
‑
mofs
‑
p20材料。
[0030]
图4:β
‑
cd
‑
mofs
‑
p50的x
‑
射线光电子能谱全谱图;
[0031]
图5:β
‑
cd
‑
mofs
‑
p50的x
‑
射线光电子能谱碳元素能谱图;
[0032]
图6:β
‑
cd
‑
mofs
‑
p50的x
‑
射线光电子能谱氧元素能谱图;
[0033]
图7:β
‑
cd
‑
mofs
‑
p50的x
‑
射线光电子能谱钾元素能谱图;
[0034]
图8:β
‑
cd
‑
mofs
‑
p20的x
‑
射线光电子能谱全谱图;
[0035]
图9:β
‑
cd
‑
mofs
‑
p20的x
‑
射线光电子能谱碳元素能谱图;
[0036]
图10:β
‑
cd
‑
mofs
‑
p20的x
‑
射线光电子能谱氧元素能谱图;
[0037]
图11:β
‑
cd
‑
mofs
‑
p20的x
‑
射线光电子能谱钾元素能谱图;
[0038]
图12:全多孔球形硅胶裸珠放大50000倍的扫描电镜图;
[0039]
图13:hplc柱1填料放大50000倍的扫描电镜图;
[0040]
图14:hplc柱2填料放大50000倍的扫描电镜图;
[0041]
图15:噁唑禾草灵消旋体拆分hplc图;
[0042]
图16:溶剂甲醇的hplc图(噁唑禾草灵拆分体系);
[0043]
图17:r
‑
噁唑禾草灵的hplc图;
[0044]
图18:马来酸氯苯那敏两对映体hplc拆分图;
[0045]
图19:氟雷拉纳两对映体hplc拆分图;
[0046]
图20:溶剂乙腈的hplc图;
[0047]
图21:雷诺嗪两对映体hplc拆分图;
[0048]
图22:溶剂水的hplc图;
[0049]
图23:羟丙哌嗪两对映体hplc拆分图;
[0050]
图24:溶剂水的hplc图;
[0051]
图25:联苯菊酯两对映体hplc拆分图;
[0052]
图26:溶剂乙腈的hplc图;
[0053]
图27:喹禾灵两对映体hplc拆分图;
[0054]
图28:溶剂水的hplc图;
[0055]
图29:奥硝唑两对映体hplc拆分图;
[0056]
图30:溶剂水的hplc图;
[0057]
图31:多沙唑嗪两对映体hplc拆分图;
[0058]
图32:溶剂水的hplc图。
具体实施方式
[0059]
下面申请人将结合具体的实施例对本发明的新手性hplc柱的制备和应用过程做详细说明,目的在于使本领域技术人员对本发明有更进一步的理解。
[0060]
实施例1、2中,所用主要仪器与试剂分别为:
[0061]
cary 5000型紫外
‑
可见
‑
近红外分光光度计(uv
‑
vis drs,安捷伦,美国);nexus
‑
470型傅立叶变换红外光谱仪(ft
‑
ir,nicolet,美国);x射线衍射仪(xrd,advance,美国);avance iii 400mhz型核磁共振波谱仪(nmr,bruker,瑞士);vg multilab 2000型光电子能谱仪(美国);jsm
‑
6700f型场发射扫描电子显微镜(fesem,日本);电磁搅拌器(上海伟业仪器厂,中国)。
[0062]
苯甲酸、氯磺酸、β
‑
环糊精、氯化钾、甲醇、十六烷基三甲基溴化铵、n,n
‑
二甲基甲
酰胺均为分析纯,国药集团化学试剂有限公司;3
‑
缩水甘油基氧丙基三甲氧基硅烷(分析纯,阿拉丁试剂);全多孔球形硅胶(5μm,比表面300mm2·
g
‑1,孔径赛分科技有限公司),间羧基苯磺酰氯为申请人根据现有技术自制。
[0063]
实施例1:β
‑
cd
‑
mofs材料β
‑
cd
‑
mofs
‑
p50及β
‑
cd
‑
mofs
‑
p20的制备
[0064]
采用一锅法合成β
‑
cd
‑
mofs
‑
p50:
[0065]
将β
‑
cd(0.67g)、间羧基苯磺酰氯(0.52g)和氯化钾(0.31g)搅拌溶解在26ml甲醇水溶液(浓度在20
‑
90v/v%均可,对产物形貌影响不大,本实施例采用浓度为60v/v%的甲醇水溶液)中,加入120mg ctab,封口,室温下,静置24h得到无色晶体,过滤,干燥备用。此时,所得β
‑
cd
‑
mofs材料的颗粒比较均匀,粒径约为50μm,命名为β
‑
cd
‑
mofs
‑
p50,其具体形貌如图1所示。
[0066]
利用蒸汽扩散法合成β
‑
cd
‑
mofs
‑
p20:
[0067]
将β
‑
cd(0.1000g)、间羧基苯磺酰氯(0.0778g)和氯化钾(0.0460g)搅拌溶解在6ml甲醇水溶液(浓度在2
‑
30v/v%均可,对产物形貌影响不大,本实施例采用浓度为9v/v%的甲醇水溶液)中,加入120mg ctab,过滤杂质后置于大的密封容器中,水浴50℃加热反应6h,冷却后静置24h得到无色立方晶体,过滤,干燥备用。因粒径约为20μm,命名为β
‑
cd
‑
mofs
‑
p20,其具体形貌如图2所示。
[0068]
用xrd等手段确定目标化合物:
[0069]
1、x
‑
射线粉末衍射图(xrd)
[0070]
如图3所示。由图3可知,β
‑
cd
‑
mofs
‑
p50在6.7
°
,9.6
°
,12.8
°
,13.5
°
,18.3
°
,18.9
°
处有明显的衍射峰,β
‑
cd
‑
mofs
‑
p20在6.2
°
,8.9
°
,10.5
°
,12.4
°
,17
°
,17.6
°
处有明显衍射峰,且与间羧基苯磺酰的衍射峰有显著的不同,与β
‑
cd的衍射峰既有相似之处又有不同,说明生成的晶体保留了β
‑
cd的骨架。表明β
‑
cd
‑
mofs
‑
p50和β
‑
cd
‑
mofs
‑
p20成功合成。
[0071]
2、x
‑
射线光电子能谱(xps)
[0072]
分别对β
‑
cd
‑
mofs
‑
p50、β
‑
cd
‑
mofs
‑
p20进行x
‑
射线光电子能谱表征,得图4,图5,图6,图7,图8,图9,图10,图11。表面元素含量如表1所示。由图4和表1所示,β
‑
cd
‑
mofs
‑
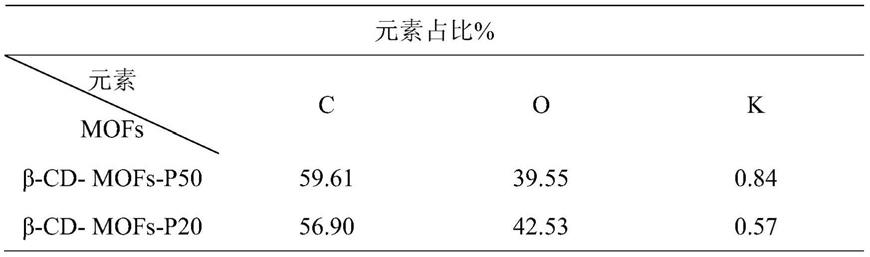
p50的c1s的结合能为286.45ev,表面含量占比为59.61%;o1s的结合能为532.86ev,表面含量占比为39.55%;k2p的结合能为293.03ev,表面含量占比为0.84%。β
‑
cd
‑
mofs
‑
p50的c1s的结合能为286.42ev,表面含量占比为56.90%;o1s的结合能为532.69ev,表面含量占比为42.53%;k2p的结合能为293.02ev,表面含量占比为0.57%。表明β
‑
cd
‑
mofs
‑
p50和β
‑
cd
‑
mofs
‑
p20均已合成,且证明金属钾存在于两种mofs材料中。
[0073]
表1表面元素含量
[0074][0075]
实施例2β
‑
cd
‑
mofs手性hplc柱的制备
[0076]
用实施例1合成的两种β
‑
cd
‑
mofs材料作为手性分离材料,分别制备成手性hplc柱填料,进而制备高效液相色谱hplc柱,用透射电镜和扫描电镜,可确认填料制备的优劣。
[0077]
1、高效液相色谱柱填料的制备方法
[0078]
制备方法为:先将商用粒径5μm的硅珠(即:全多孔球形硅胶)酸化,干燥后备用;β
‑
cd
‑
mofs(β
‑
cd
‑
mofs
‑
p50或β
‑
cd
‑
mofs
‑
p20)在交联剂γ
‑
(2,3
‑
环氧丙氧基)丙基三甲氧基硅烷(β
‑
cd
‑
mofs与交联剂摩尔比10:1)存在下、n2氛围中,加入已处理好的全多孔球形硅胶(本实施例β
‑
cd
‑
mofs与全多孔球形硅胶质量比为1:2),115℃条件下搅拌反应24h,反应完全后过滤、洗涤、干燥,备用。
[0079]
2、手性hplc柱填料的表征
[0080]
把制备好的手性hplc柱填料和裸珠用场发射扫描电镜(zeiss sigma)扫描表征,扫描结果如图12、图13、图14所示。
[0081]
由图可知,柱1(β
‑
cd
‑
mofs
‑
p50修饰)和柱2(β
‑
cd
‑
mofs
‑
p20修饰)经放大后可明显观察到表面比较粗糙且蓬松,在相同的放大倍数下发现,裸珠表面较柱1和柱2表面光滑。这表明β
‑
cd
‑
mofs
‑
p50和β
‑
cd
‑
mofs
‑
p20均已成功键合在全多孔球形硅胶上。
[0082]
3、固定相的装填
[0083]
采用匀浆法进行装填,高压泵一次填装进各种规格液相色谱不锈钢空柱当中,冲洗,甲醇饱和。
[0084]
实施例3用实施例2制备的β
‑
cd
‑
mofs手性hplc柱分离手性物质
[0085]
将β
‑
cd
‑
mofs制备成手性固定相填料,进而装填为高效液相色谱柱用于手性物质的拆分,可建立分离噁唑禾草灵、氟雷拉纳、喹禾灵、联苯菊酯、雷诺嗪、奥硝唑、多沙唑嗪、羟丙哌嗪和马来酸氯苯那敏9种手性对映体拆分和定量分析方法,还可通过该手性柱制备获得噁唑禾草灵和马来酸氯苯那敏单一对映体,据此可建立多种手性物质单一对映体hplc定量测定新方法。
[0086]
1、主要仪器及试剂
[0087]
thermo ultimate 3000(赛默飞世尔科技(中国)有限公司),lc
‑
15c岛津高效液相色谱仪(岛津企业管理(中国)有限公司),依利特液相色谱仪(分析型p230ⅱ,制备型p270);紫外检测器(分析型uv230ⅱ,制备型uv230+),as3120超声波清洗器(宁波科生仪器厂),zd
‑
2型酸度计(上海伟业仪器厂),cj
‑
1型电磁搅拌器(上海华光仪器仪表厂),溶剂过滤器(上海津腾有限公司),0.2μm微孔滤膜(上海市新亚净化器件厂),100μl微量进样器(岛津企业管理(中国)有限公司),bp211d电子分析天平(sartorius),乙腈(ar,国药集团化学试剂有限公司),磷酸二氢钠(国药集团化学试剂有限公司),三乙胺(国药集团化学试剂有限公司),超纯水(美国moleculer超纯水机生产),甲醇,乙酸,乙酸铵、柠檬酸(分析纯,国药集团);噁唑禾草灵,喹禾灵(分析标准品,百灵威科技有限公司);马来酸氯苯那敏(分析标准品,中国食品药品检定研究院);氟雷拉纳,联苯菊酯(分析标准品,上海源叶生物科技有限公司)。雷诺嗪,羟丙哌嗪,多沙唑嗪(分析标准品,上海源叶生物科技有限公司);奥硝唑(分析标准品,国药集团)。
[0088]
2、噁唑禾草灵的分离
[0089]
采用按照实施例2制备的β
‑
cd
‑
mofs手性hplc柱2(β
‑
cd
‑
mofs
‑
p20修饰,柱尺寸150mm
×
φ4.6mm),用hplc仪对噁唑禾草灵进行了手性拆分,在最佳拆分条件(乙酸铵
‑
三乙
胺缓冲溶液浓度20mmol/l,ph7.40,流动相:乙酸铵
‑
三乙胺缓冲溶液/乙腈配比53/47,温度25℃,检测波长254nm,流速0.5ml/min)下,分别测定噁唑禾草灵消旋体(4.92
×
10
‑5mol/l)、溶剂甲醇、r
‑
噁唑禾草灵(3.38
×
10
‑5mol/l),结果如图15、图16和图17所示。噁唑禾草灵两对映体分离度达8.69,结果表明噁唑禾草灵消旋体浓度在6.20
×
10
‑6mol/l~2.99
×
10
‑4mol/l范围内,r
‑
噁唑禾草灵和s
‑
噁唑禾草灵两对映体峰高和峰面积均与浓度呈较优的线性相关性(r
‑
噁唑禾草灵峰高、峰面积与浓度,s
‑
噁唑禾草灵峰高、峰面积与浓度,r分别为0.99669、0.99992、0.99619和0.99955)。重现性考察(n=7)结果显示各项色谱参数(r,s
‑
噁唑禾草灵两对映体保留时间、两色谱峰峰面积、两色谱峰峰高、两对映体分离度)的rsd均小于2%。
[0090]
3、马来酸氯苯那敏的分离
[0091]
采用按照实施例2制备的β
‑
cd
‑
mofs手性hplc柱2(β
‑
cd
‑
mofs
‑
p20修饰,柱尺寸150mm
×
φ4.6mm),用hplc仪对马来酸氯苯那敏进行了手性拆分,在最佳拆分条件(流动相磷酸
‑
三乙胺缓冲液/乙腈配比27/73,磷酸
‑
三乙胺缓冲液浓度15mmol/l,ph3.50;流速0.5ml/min;检测波长240nm)下,马来酸氯苯那敏消旋体(1.30
×
10
‑3mol/l,溶剂为超纯水,进样体积20μl)结果如图18所示。马来酸氯苯那敏两对映体达到了基线以上的分离。
[0092]
4、氟雷拉纳的分离
[0093]
在柱2(β
‑
cd
‑
mofs
‑
p20修饰,柱尺寸150mm
×
φ4.6mm)拆分氟雷拉纳对映体最佳色谱条件(柱温25℃、检测波长254nm、乙酸三乙胺缓冲液浓度5mmol/l,ph4.00、流动相配比乙酸三乙胺缓冲液/甲醇61/39、流速0.6ml/min,氟雷拉纳消旋体浓度4.84
×
10
‑4mol/l、进样体积20μl)下:分离度r
s
达1.67,拆分结果如图19、图20(溶剂乙腈)所示。由图所示可知溶剂乙腈与氟雷拉纳对映体前峰存在叠加,但相对来说溶剂响应值的影响相对较小,可以扣除。氟雷拉纳消旋体浓度在9.70
×
10
‑5mol/l~9.67
×
10
‑4mol/l范围内两个对映体的峰高和峰面积均与浓度呈现较佳的线性相关性(r分别为0.99999、0.99967、0.99845和0.98639)。重现性考察(n=7)显示各项色谱参数(氟雷拉纳两对映体保留时间、两色谱峰峰面积、两色谱峰峰高、两对映体分离度)的rsd均小于5%;
[0094]
5、雷诺嗪的分离
[0095]
柱2(β
‑
cd
‑
mofs
‑
p20修饰,柱尺寸150mm
×
φ4.6mm)在拆分雷诺嗪的最佳色谱条件(25℃,检测波长240nm,乙酸
‑
乙酸铵浓度20mmol/l,ph4.50,流动相乙酸
‑
乙酸铵:乙腈体积比=59:41,流速0.5ml/min,雷诺嗪消旋体浓度1.75
×
10
‑4mol/l,进样体积20μl)下,雷诺嗪两对映体的分离度r
s
为1.53,如图21、图22所示溶剂水对雷诺嗪对映体分离的影响较小。消旋体在3.0
×
10
‑5mol/l~3.5
×
10
‑4mol/l浓度范围内,雷诺嗪两对映体的峰高和峰面积均与浓度呈现较佳的线性相关性(r分别为:0.99936、0.98542、0.99154和0.99965)。重现性考察(n=7)显示各项色谱参数(雷诺嗪两对映体保留时间、两峰峰面积、两峰峰高、两对映体分离度)的rsd均小于2%。
[0096]
6、羟丙哌嗪的分离
[0097]
柱2(β
‑
cd
‑
mofs
‑
p20修饰,柱尺寸150mm
×
φ4.6mm)在拆分羟丙哌嗪的最佳色谱条件(25℃,检测波长240nm,乙酸
‑
乙酸铵缓冲液浓度20mmol/l,ph4.40,流动相乙酸
‑
乙酸铵缓冲液:甲醇体积比=6:94,流速0.5ml/min,进样体积20μl)下,羟丙哌嗪两对映体的分离度r
s
为1.19,羟丙哌嗪消旋体(1.07
×
10
‑4mol/l)拆分具体分离结果和溶剂水的hplc图如图
23,图24所示。
[0098]
7、联苯菊酯的分离
[0099]
柱2(β
‑
cd
‑
mofs
‑
p20修饰,柱尺寸150mm
×
φ4.6mm)在拆分联苯菊酯的最佳色谱条件(25℃,检测波长240nm,缓冲溶液柠檬酸三乙胺浓度20mm/l、ph7.00、流速0.5ml/min、流动相柠檬酸三乙胺缓冲溶液与乙腈体积比v
柠檬酸三乙胺
/v
乙腈
为5/95、进样体积20μl)下,联苯菊酯两对映体的分离度r
s
为1.06,联苯菊酯消旋体浓度5.76
×
10
‑5mol/l具体分离结果和溶剂乙腈的hplc图如图25,图26所示。
[0100]
8、喹禾灵的分离
[0101]
柱2(β
‑
cd
‑
mofs
‑
p20修饰,柱尺寸150mm
×
φ4.6mm)在拆分喹禾灵的最佳色谱条件(25℃,检测波长240nm,缓冲溶液柠檬酸三乙胺浓度20mm/l、ph4.00、流速0.5ml/min、流动相柠檬酸三乙胺缓冲溶液与甲醇体积比v
柠檬酸三乙胺
/v
甲醇
为45/55、进样体积20μl)下,喹禾灵两对映体的分离度r
s
为1.88,1.61
×
10
‑5mol/l喹禾灵消旋体拆分具体分离结果和溶剂水的hplc图如图27,图28所示;
[0102]
9、奥硝唑的分离
[0103]
以柱1(β
‑
cd
‑
mofs
‑
p50修饰,柱尺寸150mm
×
φ4.6mm)为hplc色谱柱,拆分奥硝唑对映体。在柱温25℃、检测波长240nm、缓冲溶液乙酸三乙胺(浓度20mm/l,ph4.80)、缓冲溶液乙酸三乙胺/甲醇配比98/2、流速1ml/min、奥硝唑浓度2.12
×
10
‑4mol/l、进样体积20μl的条件下,柱1可实现对奥硝唑两对映体的基线拆分,分离度r
s
为1.90。如图29、图30所示,溶剂水对分离无影响。为手性奥硝唑建立了一种新的hplc拆分方法。
[0104]
10、多沙唑嗪的分离
[0105]
在柱1(β
‑
cd
‑
mofs
‑
p50修饰,柱尺寸150mm
×
φ4.6mm)为hplc色谱柱,拆分多沙唑嗪对映体最佳色谱条件(柱温25℃、检测波长254nm、乙酸三乙胺缓冲溶液浓度20mmol/l、ph3.90、乙酸三乙胺缓冲溶液/甲醇配比29/71,进样体积20μl)下测试溶剂水样,将其色谱图与最佳色谱条件下多沙唑嗪色谱图对比,结果如图31、图32。由图可知,多沙唑嗪(消旋体浓度6.86
×
10
‑5mol/l)两对映体分离度r
s
达1.51,溶剂水的影响相对较小。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1