用于分离单甲基烃类的吸附剂及其制备和应用的制作方法
1.本发明涉及支链烃类的分离技术,特别是一种用于分离单甲基烃类的吸附剂及其制备和应用。
背景技术:
2.混合烃类成分复杂,如石脑油等由正构烷烃、异构烷烃、环烷烃及芳烃等多种烃类的混合物组成。对于蒸汽裂解乙烯而言,不同种类的烃对乙烯生成的贡献大小不同。正构烷烃的乙烯收率最高,其次是环烷烃、异构烷烃,而芳烃对乙烯生成无贡献。另一方面,作为催化重整原料,正构烷烃环化脱氢成芳烃的反应速率很慢,转化率低。多支链烷烃辛烷值高,如c5-c8多支链烷烃是优质的环保型高辛烷值汽油。
3.单支链烃,如单甲基非环状链烯和烷烃,可自身作为产品,或通过烷基化或氧化生成出不同的化学品。单甲基烃类可用作洗涤剂生产的基础化工原料,尤其是烷基苯类洗涤剂。烷基苯洗涤剂的品质特性取决于侧链烷基的化学结构。直链烷基有利于增强洗涤剂的生物降解性。洗涤剂的其它特性如在硬水中使用的有效性也受其组成和侧链结构的影响。研究测试结果显示侧链为单甲基的支链烷烃是最有效的洗涤剂前驱体。因此,精细分离混合烷烃中的直链烷烃、单支链烷烃(如单甲基的支链烷烃)、多支链烷烃等是亟待解决的提高混合烃类综合利用并提高其附加值的最有效的方法。
4.专利cn11055906a、cn1280977a、cn1749226a公开了一种疏水硅沸石的制备方法,其中采用整体转晶技术制备无粘结剂硅沸石。这种无粘结剂硅沸石在液相条件下对对二甲苯的吸附很强,对乙基苯的吸附较弱,对邻二甲苯的吸附很弱,间二甲苯几乎不被吸附,用于c8芳烃分离,由于全硅沸石孔道较小,不利于单甲基支链烷烃分离。
技术实现要素:
5.本发明所要解决的技术问题之一是克服现有技术中存在的烃类分离中只能分离正构烃类与异构烃类的技术局限性,及其无法实现混合烃类高附加值精细化利用的问题,提供一种用于分离单甲基烃类的吸附剂。本发明要解决的技术问题之二是提供一种上述吸附剂的制备方法。本发明要解决的技术问题之三是提供一种上述吸附剂在分离单甲基烃类中的应用。
6.本发明利用多孔晶体所具有的孔道择形效应,利用不同拓扑结构多孔晶体孔径不同,并利用相同拓扑结构多孔晶体的组成变化微调孔径,采用精细分离技术实现去除正构烃类后混合烃中单甲基烃类与多支链烃类的分离,实现混合烃类的有效高附加值精细化利用。
7.为解决上述技术问题,本发明第一方面在于提供一种用于分离单甲基烃类的吸附剂,所述吸附剂硅铝原子比为100到1000,优选200到800,最优选为200到500。所述吸附剂由吸附剂原粉的混合粉体构成,所述混合粉体中晶粒尺寸50-200纳米粉体的质量百分比为30%-75%,优选50%-70%,200-600纳米粉体的质量百分比为15%-45%,优选20%-30%,
600-1000纳米粉体的质量百分比为5%-30%,优选10%-20%。
8.所述混合粉体优选10元环沸石分子筛,如mel,mfi,sff,stf,nes或ter结构的分子筛中的一种或多种,更优选mfi和/或mel结构的分子筛。
9.所述吸附剂对于单甲基支链烃20℃静态吸附容量≥0.09克/克吸附剂,优选≥0.11克/克。
10.所述吸附剂,对于单甲基支链烃的静态初始5分钟吸附速率≥0.15克/分钟
·
克吸附剂,优选≥0.16克/分钟
·
克吸附剂。
11.为解决上述技术问题之二,本发明提供了一种前述吸附剂的制备方法,包括如下步骤:
12.(1)准备吸附剂原粉;
13.(2)采用滚球成型工艺,将吸附剂原粉滚球成型得到吸附剂前驱体;
14.(3)对吸附剂前驱体进行转晶处理得到成品吸附剂。
15.上述技术方案中,步骤(1)中吸附剂原粉为混合粉体,其中晶粒尺寸50-200纳米粉体以质量百分比计为30%-75%,200-600纳米粉体以质量百分比计为15%-45%,600-1000纳米粉体以质量百分比计为5%-30%。
16.上述技术方案中,步骤(2)滚球成型工艺中优选使用粘结剂,粘结剂采用水玻璃、硅溶胶、高岭土等中的至少一种,粘结剂用量以重量百分比计为粉体重量的5%-20%。
17.步骤(2)中滚球成型后优选经干燥和焙烧处理,所述干燥温度为80℃-150℃,干燥时间为3-48小时。所述焙烧温度为400℃-850℃,焙烧时间为1-24小时。
18.上述技术方案中,步骤(3)中的转晶处理为将粘结剂转晶成无粘结剂吸附剂。转晶处理条件为:转晶温度150-220℃,转晶时间2-36小时,控制转晶液ph为9-12。转晶优选在转晶液存在下进行,转晶液为水和乙二胺混合液,其中水量为混合粉体成型小球重量的100%-500%,乙二胺为混合粉体成型小球重量的1%-10%。所述转晶优选在密闭反应釜中进行。转晶完成后,经80℃-150℃、3-48小时干燥处理,再经400℃-850℃、1-24小时焙烧处理后,即得成品吸附剂。
19.本发明第三方面在于提供一种前述吸附剂在分离单甲基烃类中的应用。可用于在馏程40℃到300℃的混合烃类中分离单甲基烃类。
20.本发明利用多孔晶体所具有的孔道择形效应,利用相同拓扑结构多孔晶体的组成变化微调孔径,可对除去正构烃的混合烃类中的单甲基烃类和多支链烃类等进行精细分离,使其物尽所用,实现对混合烃类的高附加值综合利用。
21.本发明优选采用多重粒度分布微孔晶体混合滚球成型,再粘结剂转晶,硅铝原子比控制在100到1000,优选200到800,最优选为200到500,进一步提高了分离效率,实现了吸附剂的大吸附容量,对于单甲基支链烃20℃静态吸附容量≥0.09克/克吸附剂,优选≥0.11克/克吸附剂,高吸附速率,静态初始5分钟吸附速率≥0.15克/分钟
·
克吸附剂,优选≥0.16克/分钟
·
克吸附剂。
22.上述技术方案中,制备的用于混合烃类的吸附剂成品,以馏程40℃到300℃去除正构烷烃的混合烃类为原料。原料组成:单甲基烷烃24.2%;多支链烷烃75.8%。其在固定床条件下,在150℃、1.0mpa压力下,空速0.5h-1
,以吸附塔出口单甲基支链烷烃1.0%为穿透点的动态吸附量为静态吸附量85%以上。
23.吸附剂的选择性(静态吸附)计算公式如下
[0024][0025]s单甲基/多支链
‑‑‑‑‑
单甲基支链烃选择性
[0026]c单甲基s
‑‑‑‑‑
单甲基支链烃在吸附剂中的浓度
[0027]c多支链s
‑‑‑‑‑
多支链烃在吸附剂中的浓度
[0028]c单甲基l
‑‑‑‑‑
单甲基支链烃在混合烃中的浓度
[0029]c多支链l
‑‑‑‑‑
多支链烃在混合烃中的浓度。
附图说明
[0030]
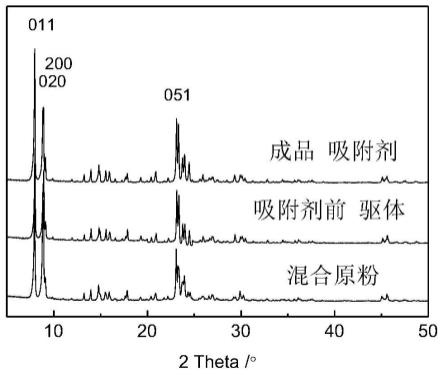
图1为实施例1中混合吸附剂原粉、吸附剂前驱体以及转晶处理后成品吸附剂的xrd图;
[0031]
图2为实施例1中吸附剂前驱体及成品吸附剂的20℃静态吸附曲线;
[0032]
图3为实施例1中成品吸附剂吸附选择性的20℃静态吸附曲线;
[0033]
图4为实施例1中成品吸附剂固定床动态吸附穿透曲线;
[0034]
图5为比较例1中吸附剂前驱体及成品吸附剂的20℃静态吸附曲线。
具体实施方式
[0035]
下面结合具体实施例对本发明作进一步阐述。但应该理解具体实施例仅为进一步阐述发明内容,本发明的保护范围并不受具体实施方式的限制。具体实施方式中所用到的分析方法如下:
[0036]
组成分析方法:
[0037]
静态吸附在20℃下进行,以均三甲苯为惰性溶剂,加入20%(重量百分比)的吸附原料,液体重量为吸附剂重量的5倍,液体与成品吸附剂混合并不断搅拌,每隔5分钟取样分析,并根据液体浓度计算单甲基烷烃与多支链烷烃的吸附量。吸附剂使用前要活化好(400℃焙烧3小时);液体组成分析采用色谱分析方法。
[0038]
色谱分析采用agilent7890a,ffap毛细管色谱柱进行分离,色谱柱采用程序升温,氢焰检测器,进样量为1μm,用面积归一化法进行定量分析。静态吸附实验是在恒温搅拌间歇式吸附装置内进行的。为使吸附剂与试剂充分接触,减少外扩散对吸附速率的影响,搅拌速度选为1000r/min。
[0039]
结晶度分析方法:
[0040]
指定混合mfi分子筛原粉结晶度为100%,吸附剂前驱体及成品(转晶)吸附剂的结晶度为混合mfi分子筛原粉mfi[011]、[020]、[002]、[051]晶面xrd峰强度加和值与成型吸附剂及成品(转晶)吸附剂晶面峰强度加和值之比。
[0041]
xrd分析采用日本理学d/max-1400型x射线衍射(xrd)仪,cu靶,kα辐射源,石墨单色器,测试电压40kv,测试电流40ma,扫描范围5~50
°
,扫描速率2
°
/min。
[0042]
【实施例1】
[0043]
吸附剂制备:
[0044]
称取硅铝原子比300、晶粒尺寸50-200纳米的mfi粉体6000克,晶粒尺寸200-600纳米的mfi粉体2500克,晶粒尺寸600-1000纳米的mfi粉体以质量百分比占1500克,均匀混合得到混合吸附剂原粉(xrd谱图见图1);
[0045]
吸附剂采用滚球成型工艺,加入粘结剂水玻璃,用量1000克,滚球成型,成型球体过筛,选取16-40目球体,经110℃干燥24小时,再经500℃焙烧处理20小时后即得吸附剂前驱体(xrd谱图见图1,20℃静态吸附曲线见图2),备用,其粉体类型及组成见表1。
[0046]
粘结剂转晶:
[0047]
首先配制转晶液:3000克纯水再加20克乙二胺,加氢氧化钠将转晶液ph调整到9.5,将转晶液及吸附剂前驱体1000克先后放入转晶釜中,密闭反应釜,轻轻搅拌1小时,并逐渐升温到180℃,转晶8小时完成,冷却后取出转晶吸附剂并用纯水洗涤到中性,110℃干燥24小时后,再经550℃焙烧处理15小时后即得成品吸附剂(xrd谱图见图1,20℃静态吸附曲线见图2),其相对结晶度见表2。
[0048]
评价:
[0049]
原料组成:单甲基烷烃24.2%;
[0050]
多支链烷烃75.8%
[0051]
原料馏程100℃到260℃
[0052]
静态评价
[0053]
静态吸附在20℃下进行,以均三甲苯为惰性溶剂,加入20%(重量百分比)的吸附原料,液体重量为吸附剂重量的5倍,液体与成品吸附剂混合并不断搅拌,每隔5分钟取样分析,并根据液体浓度计算单甲基烷烃与多支链烷烃的吸附量。
[0054]
吸附剂动态固定床评价条件
[0055]
吸附温度150℃;吸附压力1.0mpa压力下,空速0.17h-1
,以吸附塔出口单甲基支链烷烃1.0%为穿透点,考评动态吸附量指标。
[0056]
动态固定床评价结果见图4和表3。
[0057]
【实施例2】
[0058]
吸附剂制备:
[0059]
称取硅铝原子比300、晶粒尺寸50-200纳米的mel粉体6000克,晶粒尺寸200-600纳米的mel粉体2500克,晶粒尺寸600-1000纳米的mel粉体以质量百分比占1500克,均匀混合,吸附剂采用滚球成型工艺,加入粘结剂水玻璃,用量2000克,滚球成型,成型球体过筛,选取16-40目球体,经150℃干燥10小时,在经800℃焙烧处理5小时后即得吸附剂前驱体,备用,其粉体类型及组成见表1,20℃静态吸附量见表3。
[0060]
粘结剂转晶:
[0061]
首先配制转晶液:5000克纯水再加50克乙二胺,加氢氧化钠将转晶液ph调整到11,将转晶液及吸附剂前驱体1000克先后放入转晶釜中,密闭反应釜,轻轻搅拌1小时,并逐渐升温到150℃,转晶35小时完成,冷却后取出转晶吸附剂并用纯水洗涤到中性,80℃干燥45小时后,再经800℃焙烧处理5小时后即得成品吸附剂,相对结晶度见表2。吸附剂20℃静态吸附容量,静态初始5分钟吸附速率,吸附选择性见表3。
[0062]
评价:
[0063]
原料组成:单甲基烷烃24.2%;
[0064]
多支链烷烃75.8%
[0065]
原料馏程100℃到260℃
[0066]
静态评价
[0067]
静态吸附在20℃下进行,以均三甲苯为惰性溶剂,加入20%(重量百分比)的吸附原料,液体重量为吸附剂重量的5倍,液体与成品吸附剂混合并不断搅拌,每隔5分钟取样分析,并根据液体浓度计算单甲基烷烃与多支链烷烃的吸附量。
[0068]
吸附剂动态固定床评价条件
[0069]
吸附温度150℃;吸附压力1.0mpa压力下,空速0.17h-1
,以吸附塔出口单甲基支链烷烃1.0%为穿透点,考评动态吸附量指标。
[0070]
动态固定床评价结果见表3。
[0071]
【实施例3】
[0072]
吸附剂制备:
[0073]
称取硅铝原子比200、晶粒尺寸50-200纳米的mfi粉体6000克,晶粒尺寸200-600纳米的mfi粉体2500克,晶粒尺寸600-1000纳米的mfi粉体以质量百分比占1500克,均匀混合。吸附剂采用滚球成型工艺,加入粘结剂高岭土,用量1500克,滚球成型,成型球体过筛,选取16-40目球体,经80℃干燥48小时,在经600℃焙烧处理12小时后即得吸附剂前驱体,备用,其粉体类型及组成见表1,20℃静态吸附量见表3。
[0074]
粘结剂转晶:
[0075]
首先配制转晶液:1000克纯水再加100克乙二胺,加氢氧化钠将转晶液ph调整到10,将转晶液及吸附剂前驱体1000克先后放入转晶釜中,密闭反应釜,轻轻搅拌1小时,并逐渐升温到200℃,转晶20小时完成,冷却后取出转晶吸附剂并用纯水洗涤到中性,140℃干燥5小时后,再经400℃焙烧处理24小时后即得成品吸附剂(xrd谱图见图1,静态吸附曲线见图2),其相对结晶度见表2。吸附剂20℃静态吸附容量,静态初始5分钟吸附速率,吸附选择性见表3。
[0076]
评价:
[0077]
原料组成:单甲基烷烃24.2%;
[0078]
多支链烷烃75.8%
[0079]
原料馏程100℃到260℃
[0080]
静态评价
[0081]
静态吸附在20℃下进行,以均三甲苯为惰性溶剂,加入20%(重量百分比)的吸附原料,液体重量为吸附剂重量的5倍,液体与成品吸附剂混合并不断搅拌,每隔5分钟取样分析,并根据液体浓度计算单甲基烷烃与多支链烷烃的吸附量。
[0082]
吸附剂动态固定床评价条件
[0083]
吸附温度150℃;吸附压力1.0mpa压力下,空速0.17h-1
,以吸附塔出口单甲基支链烷烃1.0%为穿透点,考评动态吸附量指标。
[0084]
动态固定床评价结果见表3。
[0085]
【实施例4】
[0086]
吸附剂制备:
[0087]
称取硅铝原子比300、晶粒尺寸50-200纳米的mfi粉体7000克,晶粒尺寸200-600纳
米粉体2000克,晶粒尺寸600-1000纳米粉体以质量百分比占1000克,均匀混合。吸附剂采用滚球成型工艺,加入粘结剂水玻璃,用量500克,滚球成型,成型球体过筛,选取16-40目球体,经110℃干燥24小时,在经500℃焙烧处理18小时后即得吸附剂前驱体,备用,其粉体类型及组成见表1,20℃静态吸附量见表3。
[0088]
粘结剂转晶:
[0089]
首先配制转晶液:3000克纯水再加20克乙二胺,加氢氧化钠将转晶液ph调整到9.5,将转晶液及吸附剂前驱体1000克先后放入转晶釜中,密闭反应釜,轻轻搅拌1小时,并逐渐升温到180℃,转晶8小时完成,冷却后取出转晶吸附剂并用纯水洗涤到中性,110℃干燥24小时后,再经550℃焙烧处理15小时后即得成品吸附剂,其相对结晶度见表2。吸附剂的静态吸附容量,初始5分钟吸附速率,吸附选择性见表3。
[0090]
评价:
[0091]
原料组成:单甲基烷烃24.2%;
[0092]
多支链烷烃75.8%
[0093]
原料馏程100℃到260℃
[0094]
静态评价
[0095]
静态吸附在20℃下进行,以均三甲苯为惰性溶剂,加入20%(重量百分比)的吸附原料,液体重量为吸附剂重量的5倍,液体与成品吸附剂混合并不断搅拌,每隔5分钟取样分析,并根据液体浓度计算单甲基烷烃与多支链烷烃的吸附量。
[0096]
吸附剂动态固定床评价条件
[0097]
吸附温度150℃;吸附压力1.0mpa压力下,空速0.17h-1
,以吸附塔出口单甲基支链烷烃1.0%为穿透点,考评动态吸附量指标。
[0098]
动态固定床评价结果见表3。
[0099]
【实施例5】
[0100]
吸附剂制备:
[0101]
称取硅铝原子比200、晶粒尺寸50-200纳米的mel粉体7000克,晶粒尺寸200-600纳米的mel粉体2000克,晶粒尺寸600-1000纳米的mel粉体以质量百分比占1000克,均匀混合。吸附剂采用滚球成型工艺,加入粘结剂水玻璃,用量1000克,滚球成型,成型球体过筛,选取16-40目球体,经110℃干燥24小时,在经500℃焙烧处理20小时后即得吸附剂前驱体,备用,其粉体类型及组成见表1,20℃静态吸附量见表3。
[0102]
粘结剂转晶:
[0103]
首先配制转晶液:3000克纯水再加20克乙二胺,加氢氧化钠将转晶液ph调整到9.5,将转晶液及吸附剂前驱体1000克先后放入转晶釜中,密闭反应釜,轻轻搅拌1小时,并逐渐升温到180℃,转晶8小时完成,冷却后取出转晶吸附剂并用纯水洗涤到中性,110℃干燥24小时后,再经550℃焙烧处理15小时后即得成品吸附剂,其相对结晶度见表2。吸附剂的静态吸附容量,初始5分钟吸附速率,吸附选择性见表3。
[0104]
评价:
[0105]
原料组成:单甲基烷烃24.2%;
[0106]
多支链烷烃75.8%
[0107]
原料馏程100℃到260℃
[0108]
静态评价
[0109]
静态吸附在20℃下进行,以均三甲苯为惰性溶剂,加入20%(重量百分比)的吸附原料,液体重量为吸附剂重量的5倍,液体与成品吸附剂混合并不断搅拌,每隔5分钟取样分析,并根据液体浓度计算单甲基烷烃与多支链烷烃的吸附量。
[0110]
吸附剂动态固定床评价条件
[0111]
吸附温度150℃;吸附压力1.0mpa压力下,空速0.17h-1
,以吸附塔出口单甲基支链烷烃1.0%为穿透点,考评动态吸附量指标。
[0112]
动态固定床评价结果见表3。
[0113]
【实施例6】
[0114]
吸附剂制备:
[0115]
称取硅铝原子比500、晶粒尺寸50-200纳米的mfi粉体5000克,晶粒尺寸200-600纳米的mfi粉体3000克,晶粒尺寸600-1000纳米的mfi粉体以质量百分比占2000克,均匀混合。吸附剂采用滚球成型工艺,加入粘结剂硅溶胶,用量1000克,滚球成型,成型球体过筛,选取16-40目球体,经110℃干燥24小时,在经500℃焙烧处理20小时后即得吸附剂前驱体,备用,其粉体类型及组成见表1,20℃静态吸附量见表3。
[0116]
粘结剂转晶:
[0117]
首先配制转晶液:3000克纯水再加20克乙二胺,醋酸铵5克,加氢氧化钠将转晶液ph调整到10.0,将转晶液及吸附剂前驱体1000克先后放入转晶釜中,密闭反应釜,轻轻搅拌1小时,并逐渐升温到180℃,转晶8小时完成,冷却后取出转晶吸附剂并用纯水洗涤到中性,110℃干燥24小时后,再经550℃焙烧处理15小时后即得成品吸附剂,其相对结晶度见表2。吸附剂的20℃静态吸附容量,初始5分钟吸附速率,吸附选择性见表3。
[0118]
评价:
[0119]
原料组成:单甲基烷烃24.2%;
[0120]
多支链烷烃75.8%
[0121]
原料馏程100℃到260℃
[0122]
静态评价
[0123]
静态吸附在20℃下进行,以均三甲苯为惰性溶剂,加入20%(重量百分比)的吸附原料,液体重量为吸附剂重量的5倍,液体与成品吸附剂混合并不断搅拌,每隔5分钟取样分析,并根据液体浓度计算单甲基烷烃与多支链烷烃的吸附量。
[0124]
吸附剂动态固定床评价条件
[0125]
吸附温度150℃;吸附压力1.0mpa压力下,空速0.17h-1
,以吸附塔出口单甲基支链烷烃1.0%为穿透点,考评动态吸附量指标。
[0126]
动态固定床评价结果见表3。
[0127]
【实施例7】
[0128]
吸附剂制备:
[0129]
称取硅铝原子比900、晶粒尺寸50-200纳米的mfi粉体6000克,晶粒尺寸200-600纳米的mfi粉体2500克,晶粒尺寸600-1000纳米的mfi粉体以质量百分比占1500克,均匀混合。吸附剂采用滚球成型工艺,加入粘结剂水玻璃,用量1000克,滚球成型,成型球体过筛,选取16-40目球体,经110℃干燥24小时,在经500℃焙烧处理20小时后即得吸附剂前驱体(20℃
静态吸附曲线见图5),吸附备用,其粉体类型及组成见表1,20℃静态吸附量见表3。
[0130]
粘结剂转晶:
[0131]
首先配制转晶液:3000克纯水再加20克乙二胺,加氢氧化钠将转晶液ph调整到9.5,将转晶液及吸附剂前驱体1000克先后放入转晶釜中,密闭反应釜,轻轻搅拌1小时,并逐渐升温到180℃,转晶8小时完成,冷却后取出转晶吸附剂并用纯水洗涤到中性,110℃干燥24小时后,再经550℃焙烧处理15小时后即得成品吸附剂(20℃静态吸附曲线见图5),其相对结晶度见表2。吸附剂20℃静态吸附容量,初始5分钟吸附速率,吸附选择性见表3。
[0132]
评价:
[0133]
原料组成:单甲基烷烃24.2%;
[0134]
多支链烷烃75.8%
[0135]
原料馏程100℃到260℃
[0136]
静态评价
[0137]
静态吸附在20℃下进行,以均三甲苯为惰性溶剂,加入20%(重量百分比)的吸附原料,液体重量为吸附剂重量的5倍,液体与成品吸附剂混合并不断搅拌,每隔5分钟取样分析,并根据液体浓度计算单甲基烷烃与多支链烷烃的吸附量。
[0138]
吸附剂动态固定床评价条件
[0139]
吸附温度150℃;吸附压力1.0mpa压力下,空速0.17h-1
,以吸附塔出口单甲基支链烷烃1.0%为穿透点,考评动态吸附量指标。
[0140]
动态固定床评价结果见表3。
[0141]
【比较例1】
[0142]
吸附剂制备:
[0143]
称取硅铝原子比300、晶粒尺寸50-200纳米的mfi粉体10000克。吸附剂采用滚球成型工艺,加入粘结剂水玻璃,用量1000克,滚球成型,成型球体过筛,选取16-40目球体,经110℃干燥24小时,在经500℃焙烧处理20小时后即得吸附剂前驱体,备用,其粉体类型及组成见表1,20℃静态吸附量见表3。
[0144]
粘结剂转晶:
[0145]
首先配制转晶液:3000克纯水再加20克乙二胺,加氢氧化钠将转晶液ph调整到9.5,将转晶液及吸附剂前驱体1000克先后放入转晶釜中,密闭反应釜,轻轻搅拌1小时,并逐渐升温到180℃,转晶8小时完成,冷却后取出转晶吸附剂并用纯水洗涤到中性,110℃干燥24小时后,再经550℃焙烧处理15小时后即得成品吸附剂,其相对结晶度见表2。吸附剂20℃静态吸附容量,初始5分钟吸附速率,吸附选择性见表3。
[0146]
评价:
[0147]
原料组成:单甲基烷烃24.2%;
[0148]
多支链烷烃75.8%
[0149]
原料馏程100℃到260℃
[0150]
静态评价
[0151]
静态吸附在20℃下进行,以均三甲苯为惰性溶剂,加入20%(重量百分比)的吸附原料,液体重量为吸附剂重量的5倍,液体与成品吸附剂混合并不断搅拌,每隔5分钟取样分析,并根据液体浓度计算单甲基烷烃与多支链烷烃的吸附量。
[0152]
吸附剂动态固定床评价条件
[0153]
吸附温度150℃;吸附压力1.0mpa压力下,空速0.17h-1
,以吸附塔出口单甲基支链烷烃1.0%为穿透点,考评动态吸附量指标。
[0154]
固定床评价结果见表3。
[0155]
表1
[0156][0157]
表2
[0158][0159][0160]
*指定混合mfi分子筛原粉结晶度为100%,成型吸附剂及成品(转晶)吸附剂的结晶度为混合mfi分子筛原粉mfi[011]、[020]、[002]、[051]晶面峰强度加和值与成型吸附剂及成品(转晶)吸附剂晶面峰强度加和值之比。
[0161]
表3
[0162][0163]
*以吸附塔出口单甲基支链烷烃1.0%为穿透点计算吸附容量
[0164]
**吸附30分钟时,吸附剂中单甲基支链烷烃与多支链烷烃之比
[0165]
***动态吸附容量与静态吸附容量之比。
[0166]
由图1、表2可知转晶后成品吸附剂结晶度显著提高,其结晶度已到达原粉xrd结晶度。图2显示成品吸附剂静态吸附量明显大于吸附剂前驱体(转晶前),具体数据见表3。吸附剂20℃静态吸附容量,静态初始5分钟吸附速率,吸附选择性见图3和表3。图3显示成品吸附剂对单甲基支链烃的吸附量远大于多支链烃,表明其具有高吸附选择性,具体数值见表3。图4显示动态吸附穿透点上升曲线陡峭,表明穿透点动态饱和吸附量大,具体数值见表3。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1