一种磁性纳米双酸催化剂及其制备方法和应用
本发明涉及苯并二氮杂卓类化合物合成
技术领域:
,尤其涉及一种磁性纳米双酸催化剂及其制备方法和应用。
背景技术:
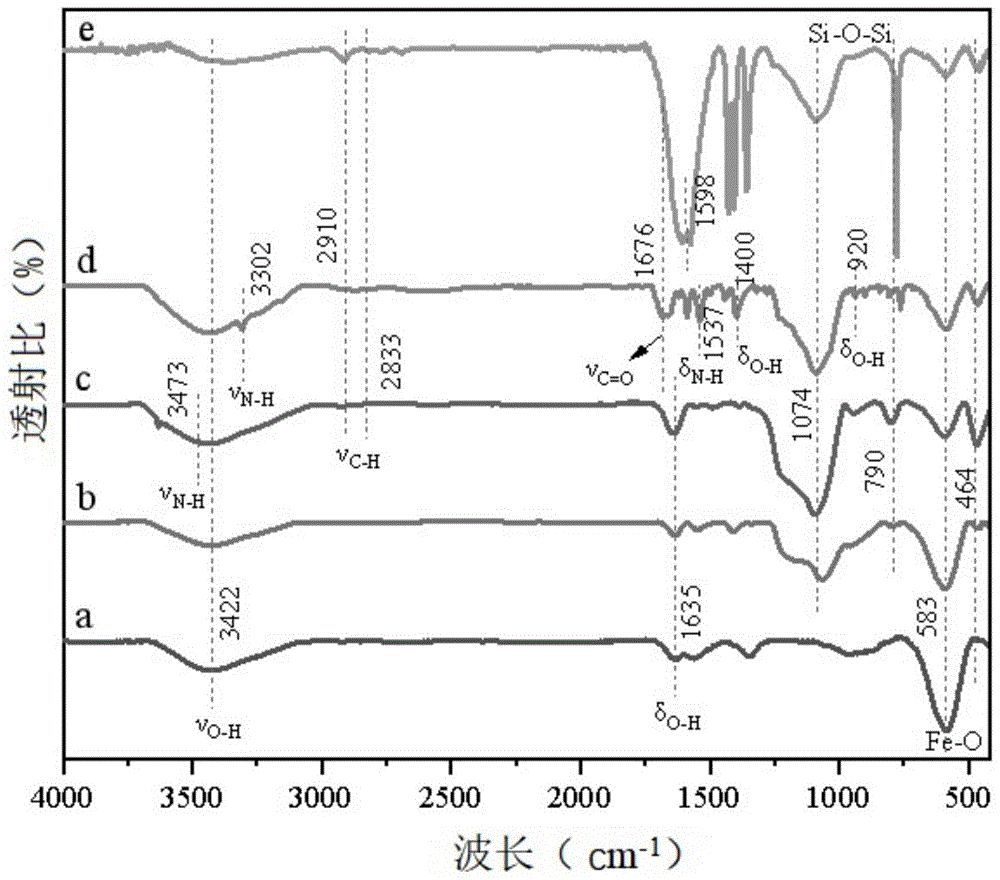
:1,5-苯并二氮杂卓类化合物是一类具有重要药理活性和生理活性的化合物,常用作抗焦虑、抗抑郁、抗惊厥、镇静、镇痛等方面的药物,此外,此类化合物还报道具有抗菌、抗病毒和拮抗等作用。除此之外,1,5-苯并二氮杂卓类化合物还是合成稠环类化合物的重要中间体。因此,研究开发1,5-苯并二氮杂卓类化合物的相关合成技术具有广阔的应用前景。目前,1,5-苯并二氮卓类化合物的典型合成方法主要是通过邻苯二胺和α,β-不饱和羰基化合物或β-卤代酮或酮类化合物间的缩合/环化来实现的。在反应过程中一般选择加入酸催化剂,如浓盐酸、冰醋酸或浓硫酸等等。但是,这些酸催化剂的选择性较差,催化效率不高且难以回收利用。目前,还有将贵金属钯纳米粒子沉积在磁性纳米粒子上作为合成1,5-苯并二氮卓类化合物的催化剂的报道。虽然,磁性纳米负载固体酸催化剂解决了回收利用的问题,但是,由于合成1,5-苯并二氮卓类化合物的反应一般涉及多步反应,因此,现有催化剂的催化选择性和催化效率依然不高,且负载催化剂的稳定性较差,导致催化剂的循环使用性较差,限制了磁性纳米负载酸催化剂在1,5-苯并二氮杂卓合成中的应用。因此,研发一种催化活性高、选择性高且稳定性较好的用于合成1,5-苯并二氮杂卓类化合物的催化剂具有十分重要的意义。技术实现要素:针对现有的合成1,5-苯并二氮杂卓类化合物的催化剂催化活性不高、选择性较差以及循环使用寿命较差的问题,本发明提供一种磁性纳米双酸催化剂及其制备方法和应用,本发明通过将lewis酸和酸通过化学键合的方式负载到钴铁氧体磁性载体上,不但显著提高了催化剂的催化活性和选择性,还显著提高了催化剂的稳定性,催化剂循环使用多次后催化活性无明显下降。为解决上述技术问题,本发明提供的技术方案是:本申请第一方面提供了一种磁性纳米双酸催化剂,包括氨基化cofe2o4@sio2核壳纳米粒子载体,以及化学键合负载在所述载体上的酸和过渡金属lewis酸;其中,所述酸的结构如式(ⅰ)所示:r1、r2、r3、r4为h、羧基或氨基,且r1、r2、r3、r4中至少有一个为羧基;所述酸中氨基邻位的羧基与所述载体表面的氨基缩合形成酰胺键;所述酰胺键中的n和所述酰胺键邻位氨基中的n与所述过渡金属lewis酸中的金属原子形成配位键。相对于现有技术,本发明选择氨基化cofe2o4@sio2核壳纳米粒子作为载体,利用氨基化cofe2o4@sio2核壳纳米粒子表面的氨基与酸中的羧基脱水缩合反应形成酰胺键,实现了酸在磁性纳米粒子表面的锚定,然后以酸为负载媒介,巧妙利用lewis酸中金属原子与酰胺键中的n以及酰胺键邻位氨基中的n配位,形成稳定的六元环结构,实现了将lewis酸稳定负载在含羧基的磁性纳米粒子表面,得到了既键合酸又结合lewis酸的双酸催化剂。本发明提供的双酸复合催化剂的催化活性高,可同时催化多种有机反应,有利于实现一锅法合成目标产物的目的,且便于从体系中分离回收,热稳定好,多次重复使用后活性无明显下降,是一种极具应用价值的新型催化剂,潜在应用领域广阔。优选的,所述酸为2-氨基对苯二甲酸。优选的酸对1,5苯并二氮杂卓类化合物的合成反应具有较高的催化效果。优选的,所述过渡金属lewis酸为三氯化铈、氯化铜、氯化镍或醋酸钯。进一步优选的,所述过渡金属lewis酸为三氯化铈。优选的lewis酸具有较高的配位能力,能与酸和氨基形成酰胺键中的n和酰胺键邻位氨基中的n形成较强的配位键,从而形成稳定的六元环结构,提高lewis酸在载体表面的稳定性,从而提高了催化剂的稳定性;除此之外,三氯化铈对1,5-苯并二氮杂卓类化合物的合成反应具有优异的催化活性,可以提高负载催化剂的催化活性。优选的,所述磁性纳米双酸催化剂的结构如式(ⅱ)所示:其中,r为甲基或乙基。本申请第二方面提供了上述磁性纳米双酸催化剂的制备方法,包括如下步骤:步骤a,制备cofe2o4@sio2核壳纳米粒子;步骤b,将所述cofe2o4@sio2核壳纳米粒子分散于第一有机溶剂中,加入含氨基的硅烷偶联剂,混合均匀,加热反应,分离产物,洗涤,干燥,得氨基化cofe2o4@sio2核壳纳米粒子;步骤c,将所述氨基化cofe2o4@sio2核壳纳米粒子和所述酸分散于第二有机溶剂中,于140-150℃反应20-24h,分离产物,洗涤,干燥,得负载酸的cofe2o4@sio2核壳纳米粒子;步骤d,将所述负载酸的cofe2o4@sio2核壳纳米粒子和所述过渡金属lewis酸分散于第三有机溶剂中,于70-78℃反应20-24h,分离产物,洗涤,干燥,得所述磁性纳米双酸催化剂。相对于现有技术,本发明首先采用二氧化硅对磁性尖晶石钴铁氧体进行包覆形成核壳结构的cofe2o4@sio2纳米粒子,不但降低了磁性纳米粒子的团聚,而且二氧化硅表面的羟基又可以为进一步氨基功能化提供反应基团;通过二氧化硅表面的oh与硅烷偶联剂中的烷氧基反应,制备得到氨基化的cofe2o4@sio2核壳磁性纳米粒子;进而利用磁性纳米粒子表面的氨基与酸的羧基脱水缩合反应,形成酰胺键,实现了酸化学键合负载在磁性纳米粒子表面;进而以形成的酰胺键为负载媒介,通过lewis酸中的金属原子与酰胺键中的n以及酰胺键邻位的n配位,形成稳定的六元环结构;最终制备得到了一种负载铈的磁性纳米双酸催化剂。本发明提供的磁性纳米双酸催化剂的制备方法,操作简单,原料易得,且制备过程中无需任何稳定剂和表面活性剂来稳定活性组分,制备所得的催化剂表面的活性中心多,反应活性高,且活性组分是以化学键合的形式负载到载体表面,稳定性好,可通过磁铁吸附分离,具有易回收和可多次重复使用的优点。优选的,步骤b中,所述cofe2o4@sio2核壳纳米粒子的质量与含氨基的硅烷偶联剂的物质的量比为1-2:5-7其中,质量的单位是克,物质的量的单位是毫摩尔。优选的,步骤b中,所述cofe2o4@sio2核壳纳米粒子与第一有机溶剂的质量体积比为1-2:50-100,其中,质量的单位是克,体积的单位是毫升。优选的,步骤c中,所述氨基化cofe2o4@sio2核壳纳米粒子与酸的质量比为1:1-1.2。优选的,步骤c中,所述氨基化cofe2o4@sio2核壳纳米粒子与第二有机溶剂的质量体积比为1:15-20,其中,质量的单位是克,体积的单位是毫升。优选的,步骤d中,所述负载酸的cofe2o4@sio2核壳纳米粒子的质量与过渡金属lewis酸的物质的量比为1:1.5-2,其中,质量的单位是克,物质的量的单位是毫摩尔。优选的,步骤d中,所述负载酸的cofe2o4@sio2核壳纳米粒子与第三有机溶剂的质量体积比为1:20-30,其中,质量的单位是克,体积的单位是毫升。优选的,步骤b中,所述含氨基的硅烷偶联剂为3-氨丙基三乙氧基硅烷或3-氨丙基三甲氧基硅烷。优选的硅烷偶联剂可与磁性纳米粒子中二氧化硅表面的羟基充分反应,实现对磁性纳米粒子的氨基功能化,从而有利于后续化学键合负载酸。优选的,步骤b中,所述第一有机溶剂为甲苯或正己烷。优选的有机溶剂有利于使硅烷偶联剂与cofe2o4@sio2核壳纳米粒子表面的羟基充分反应,实现对纳米表面的氨基功能化。优选的,步骤c中,所述第二有机溶剂为n,n-二甲基甲酰胺。优选的有机溶剂可使式(ⅰ)所示的酸充分溶解,并促进酸的羧基与cofe2o4@sio2核壳纳米粒子表面的氨基充分反应,实现酸的化学键合负载。优选的,步骤d中,所述第三有机溶剂为无水乙醇或丙酮。优选的有机溶剂可以促进过渡金属的lewis酸与酰胺中的n以及酰胺邻位氨基中的n发生配位,从而有利于形成稳定的六元环结构。优选的,所述cofe2o4@sio2核壳纳米粒子的制备方法具体包括如下步骤:将二氯化钴和三氯化铁溶于去离子水中,混合均匀,升温至80-85℃,调节ph至10-12,保温反应1-2h,分离产物,洗涤,干燥,得cofe2o4纳米颗粒;将所述cofe2o4纳米颗粒分散于乙醇水溶液中,依次加入浓氨水和正硅酸乙酯,10-30℃搅拌反应24-30h,分离产物,洗涤,干燥,得所述cofe2o4@sio2核壳纳米粒子。优选的,所述二氯化钴和三氯化铁的摩尔比为1:2。优选的,所述cofe2o4纳米颗粒与乙醇水溶液的质量体积比为1:40-80,其中,质量的单位是克,体积的单位是毫升;其中,乙醇水溶液中乙醇和水的体积比为4-5:1。优选的,所述cofe2o4纳米颗粒与浓氨水的质量体积比为1-2:1-2,其中,质量的单位是克,体积的单位是毫升。优选的,所述cofe2o4纳米颗粒与正硅酸乙酯的质量体积比为1-2:1-2,其中,质量的单位是克,体积的单位是毫升。本发明中所述浓氨水均指的是浓度为25-28wt%的工业氨水。上述制备过程中分离产物的方法均选择以磁铁吸附分离的方法。本申请第三方面还提供了上述任一项所述的磁性纳米双酸催化剂在合成1,5-苯并二氮杂卓类化合物中的应用。本发明提供的磁性纳米双酸催化剂可以应用于下列反应中:当r1=h时,r2=h,ch3,cl,br,och3;当r1=ch3时,r2=ch3;n=1,2。现有技术中负载型固体催化剂大多是载体通过弱的范德华力与活性组分结合,催化剂的稳定性较差,无法实现循环使用。且目前一锅法合成1,5-苯并二氮杂卓类化合物过程涉及多个不同的有机反应,单一催化剂很难实现高选择性、高活性催化。本发明提供的双酸负载催化剂可催化不同类型的有机反应,实现了串联反应一锅绿色合成1,5-苯并二氮杂卓类化合物,且催化活性和选择性高,易于从反应体系中分离,且稳定性好,可重复使用,在合成1,5-苯并二氮杂卓类化合物领域具有广阔的应用前景。附图说明图1为本发明实施例1制备的各步反应产物的傅里叶红外光谱图(ft-ir),其中,a:cofe2o4,b:cofe2o4@sio2,c:cofe2o4@sio2@aptes,d:cofe2o4@sio2@aptes@ata,e:cofe2o4@sio2@aptes@ata@cecl3;图2为本发明实施例1制备的各步反应产物的x射线衍射图(xrd),其中,a:cofe2o4,b:cofe2o4@sio2,c:cofe2o4@sio2@aptes,d:cofe2o4@sio2@aptes@ata,e:cofe2o4@sio2@aptes@ata@cecl3;图3为本发明实施例1制备的cofe2o4和cofe2o4@sio2@aptes@ata@cecl3的扫描电镜图(sem),其中,a:cofe2o4,b:cofe2o4@sio2@aptes@ata@cecl3;图4为本发明实施例1制备的cofe2o4@sio2@aptes@ata@cecl3的x射线能量色散谱图(eds);图5为本发明实施例1制备的cofe2o4@sio2@aptes@ata@cecl3的透射电镜图(tem);图6a为本发明实施例1制备的cofe2o4@sio2@aptes@ata@cecl3的x射线光电子能谱图(xps);图6b为本发明实施例1制备的cofe2o4@sio2@aptes@ata@cecl3的fe2p电子结合能谱;图6c为本发明实施例1制备的cofe2o4@sio2@aptes@ata@cecl3的c1s电子结合能谱;图6d为本发明实施例1制备的cofe2o4@sio2@aptes@ata@cecl3的o1s电子结合能谱;图6e为本发明实施例1制备的cofe2o4@sio2@aptes@ata@cecl3的n1s电子结合能谱;图6f为本发明实施例1制备的cofe2o4@sio2@aptes@ata@cecl3的ce3d电子结合能谱;图7为本发明实施例1制备的cofe2o4@sio2@aptes@ata@cecl3的n2低温物理吸附解吸图(bet),其中,左上角插图为孔径分布示意图;图8为本发明实施例1制备的cofe2o4@sio2@aptes@ata@cecl3的磁滞回线图(vsm),其中,左上角为外加磁铁的产物实际分离效果图;图9为本发明实施例1制备的cofe2o4@sio2@aptes@ata@cecl3的循环使用性能图。具体实施方式为了使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。为了更好的说明本发明,下面通过实施例做进一步的举例说明。下述实施例中所有的试剂如无特殊说明均为现有市售试剂,实验方法如无特殊说明均采用现有的实验方法。实施例1一种磁性纳米双酸催化剂(cofe2o4@sio2@aptes@ata@cecl3)的制备方法:步骤a,cofe2o4纳米颗粒的制备:称取2.70gfecl3·6h2o和1.19gcocl2·6h2o溶解在50ml去离子水中,超声分散40min,逐渐升温至80℃,在搅拌条件下用3mol/l的naoh溶液调节反应体系的ph为10.5,于80℃保温搅拌反应1h,通过外加磁铁分离cofe2o4纳米颗粒,然后依次用去离子水和无水乙醇洗涤3次,在80℃真空干燥24h,得cofe2o4纳米颗粒。步骤b,制备cofe2o4@sio2磁性纳米颗粒:称取1g上述制备的cofe2o4纳米颗粒,加入40ml乙醇和10ml去离子水的混合溶液中,超声分散30min,然后加入1ml浓氨水,室温搅拌30min后,滴加1ml正硅酸乙酯,室温搅拌反应24h,用外加磁铁分离产物,并用去离子水和无水乙醇洗涤3次,在80℃真空干燥24h,得cofe2o4@sio2磁性纳米颗粒。步骤c,制备氨基化cofe2o4@sio2磁性纳米颗粒:称取1g上述制备的cofe2o4@sio2磁性纳米颗粒和5mmol的3-氨丙基三乙氧基硅烷,加入50ml甲苯中,于110℃回流反应24h,冷却至室温,用外加磁铁分离产物,并用去离子水和无水乙醇洗涤3次,在80℃真空干燥24h,得氨基化cofe2o4@sio2磁性纳米颗粒(cofe2o4@sio2@aptes)。步骤d,制备负载酸的cofe2o4@sio2核壳纳米粒子:称取1g制备的cofe2o4@sio2@aptes加入20mln,n-二甲基甲酰胺中,超声分散30min,然后加入1g2-氨基对苯二甲酸(ata),加热至145℃反应24h,冷却至室温,用外加磁铁分离产物,并用去离子水和无水乙醇洗涤3次,在80℃真空干燥24h,得负载酸的cofe2o4@sio2核壳纳米粒子(cofe2o4@sio2@aptes@ata)。步骤e,制备双酸催化剂:称取1g制备的cofe2o4@sio2@aptes@ata,2mmolcecl3·7h2o加入20ml无水乙醇中,超声分散30min,然后加热至75℃搅拌反应20h,冷却至室温,用外加磁铁分离产物,并用去离子水和无水乙醇洗涤3次,在80℃真空干燥24h,得双酸催化剂(cofe2o4@sio2@aptes@ata@cecl3)。具体反应方程式如下:图1为本实施例各步骤制备的反应产物的傅里叶红外光谱图(ft-ir)。在在cofe2o4的磁性纳米粒子光谱中(a),位于583cm-1和464cm-1的两个峰是cofe2o4的八面体和四面体位置的铁氧伸缩振动。此外,1676cm-1处的吸收带和3422cm-1处的宽带分别是由于o-h键的弯曲振动和伸缩振动。在cofe2o4@sio2的磁性纳米粒子光谱中(b)可以看出,宽吸收带出现在1074cm-1和790cm-1,分别对应si-o-si键的弯曲、对称和不对称振动。在cofe2o4@sio2@aptes的磁性纳米粒子光谱中(c)可以看出,在2833cm-1和2910cm-1处的两个峰,是硅烷偶联剂中c-h的伸缩振动。此外,在3473cm-1处出现的峰是氨基中n-h键的伸缩振动,说明硅烷偶联剂成功固定在磁性纳米粒子上。在cofe2o4@sio2@aptes@ata的磁性纳米粒子光谱中(d)可以看出,在3302cm-1和1537cm-1处出现与仲酰胺的n-h相关的峰和在1676cm-1处出现可归因于仲酰胺的c-o键的峰,在1400cm-1和920cm-1处出现的两个弱吸收带是羧基的o-h键的弯曲振动,证实了2-氨基对苯二甲酸(ata)在磁性纳米粒子上的官能化。在cofe2o4@sio2@aptes@ata@cecl3的磁性纳米粒子光谱中(e)可以看出,c=o的带移至1598cm-1的较低频率,证实了铈络合物成功地固定在cofe2o4@sio2@aptes@ata磁性纳米粒子的表面上。图2为本实施例各步骤制备的反应产物的x射线衍射图(xrd)。在18.3°(111晶面)、30.2°(220晶面)、35.6°(311晶面)、43.3°(400晶面)、53.6°(422晶面)、57.3°(511晶面)、62.6°(440晶面)的2θ值的衍射峰为纯cofe2o4相(jcpdspdf#221086)立方尖晶石结构。基于b-e所示的xrd图案,出现了与cofe2o4相似的强峰和图案,证实了有机分子对cofe2o4的表面修饰不会引起相变,从而证实了cofe2o4的晶体结构在官能化过程中保持完整。图3为本实施例制备的cofe2o4纳米颗粒和cofe2o4@sio2@aptes@ata@cecl3纳米颗粒的扫描电镜图(sem)。从图中可以看出,本实施例制备的cofe2o4是平均尺寸约为36nm的均匀的球形颗粒,cofe2o4@sio2@aptes@ata@cecl3是平均尺寸约为40nm的准球形颗粒。这表明由于表面改性导致最终cofe2o4@sio2@aptes@ata@cecl3纳米粒子的尺寸比纯的cofe2o4大,并且纳米粒子的结构没有发生变化和损伤。图4是本实施例制备的cofe2o4@sio2@aptes@ata@cecl3纳米颗粒的x射线能量色散谱图(eds)。通过能谱仪检测了所有预期的主要元素,包括c,n,o,si,fe,co,ce。基于这些结果,可以合理地证明三氯化铈已经成功地与2-氨基对苯二甲酸(ata)配位。图5是本实施例制备的cofe2o4@sio2@aptes@ata@cecl3纳米颗粒的透射电镜图(tem)。本实施例制备的cofe2o4@sio2@aptes@ata@cecl3磁性纳米粒子可被视为球形核壳纳米粒子,其是由均匀分散的无定形二氧化硅包覆的暗磁铁矿核。此外,磁性纳米粒子的平均尺寸为40-45nm,二氧化硅包覆层的厚度为4-9nm。同时,由于二氧化硅覆盖的厚度,磁性纳米粒子中出现了一些聚集现象。图6是本实施例制备的cofe2o4@sio2@aptes@ata@cecl3纳米颗粒的x射线光电子能谱图(xps)。图6acofe2o4@sio2@aptes@ata@cecl3纳米催化剂的全谱证实了铁、钴、碳、氧、氮、硅、氯和铈元素的共存,这与eds的结果一致。图6b显示了结合能为711.5ev(fe2p3/2)和723.9ev(fe2p1/2)的峰,证实了cofe2o4中fe3+的存在。如图6c所示,在283.8ev、283.9ev、284.6ev、285.7ev、287.6ev和288.5ev的峰分别是c-si、c-h、c-c/c=c、c-n、o=c-nh和o=c-o中各个化学环境中c1s的结合能。cofe2o4@sio2@aptes@ata@cecl3中的c1s含有o=c-nh的峰,进一步证实了ata和aptes的连接是以化学键的形式存在的。cofe2o4@sio2@aptes@ata@cecl3纳米催化剂的o1s光谱显示在图6d中,显示在529.9ev、531.2ev、531.9ev、533.1ev和534.5ev处的峰分别对应于fe-o、o=c-nh、si-o、o=c-o和co-o。cofe2o4@sio2@aptes@ata@cecl3纳米催化剂在399.5ev和400.6ev的峰分别是c-n和o=c-nh引起的n1s的结合能(图6e)。图6f中885.9ev和904.4ev的峰是ce3d5/2和ce3d3/2的结合能。ce3d峰的出现是ce(ⅲ)负载在cofe2o4@sio2@aptes@ata@cecl3纳米催化剂表面的最有效证据。图7为本实施例制备的cofe2o4@sio2@aptes@ata@cecl3纳米颗粒的n2低温物理吸附解吸图(bet)。根据iupac的分类,cofe2o4@sio2@aptes@ata@cecl3纳米颗粒的吸附等温线属于ⅳ型,具有明显的h1磁滞回线,表明磁性纳米粒子具有介孔特性。磁性纳米粒子的比表面积、总孔容和平均孔径分别为37.19m2·g-1、0.16cc·g-1和7.10nm。图8是本实施例制备的各步骤制备的反应产物的磁滞回线图(vsm)。结果表明,cofe2o4、cofe2o4@sio2@aptes@ata、cofe2o4@sio2@aptes@ata@cecl3的饱和磁化强度值分别为57.91emu/g、44.70emu/g、35.66emu/g。对于三个样品,在没有剩磁或矫顽力的情况下,磁化曲线随着使用的磁场的增加急剧增加,证明了这些纳米颗粒的超顺磁性特征。由于磁性纳米粒子表面接枝了功能化的基团,cofe2o4@sio2@aptes@ata和cofe2o4@sio2@aptes@ata@cecl3的磁化饱和值低于cofe2o4。但如图8的插图所示,它们仍然可以用外部磁场有效和容易地从反应混合物中分离。实施例2本实施例提供一种磁性纳米双酸催化剂(cofe2o4@sio2@aptes@ata@cucl2)的制备方法,其中步骤a-步骤d与实施例1完全相同,步骤e具体为:称取1g制备的cofe2o4@sio2@aptes@ata,2mmolcucl2·2h2o加入20ml无水乙醇中,超声分散30min,然后加热至75℃搅拌反应20h,冷却至室温,用外加磁铁分离产物,并用去离子水和无水乙醇洗涤3次,在80℃真空干燥24h,得双酸催化剂(cofe2o4@sio2@aptes@ata@cucl2)。实施例3本实施例提供一种磁性纳米双酸催化剂(cofe2o4@sio2@aptes@ata@nicl2)的制备方法,其中步骤a-步骤d与实施例1完全相同,步骤e具体为:称取1g制备的cofe2o4@sio2@aptes@ata,2mmolnicl2·6h2o加入20ml无水乙醇中,超声分散30min,然后加热至75℃搅拌反应20h,冷却至室温,用外加磁铁分离产物,并用去离子水和无水乙醇洗涤3次,在80℃真空干燥24h,得双酸催化剂(cofe2o4@sio2@aptes@ata@nicl2)。实施例4本实施例提供一种磁性纳米双酸催化剂(cofe2o4@sio2@aptes@ata@pd(oac)2)的制备方法,其中步骤a-步骤d与实施例1完全相同,步骤e具体为:称取1g制备的cofe2o4@sio2@aptes@ata,2mmolpd(oac)2加入20ml丙酮中,超声分散30min,然后加热至75℃搅拌反应20h,冷却至室温,用外加磁铁分离产物,并用丙酮洗涤3次,在80℃真空干燥24h,得双酸催化剂(cofe2o4@sio2@aptes@ata@pd(oac)2)。催化活性评价:实施例1-4制备的双酸催化剂可应用于作为如下一类反应的催化剂:当r1=h时,r2=h,-ch3,cl,br,-och3;当r1=-ch3时,r2=-ch3;n=1,2。下面以制备2-苄基-3酯基-4-甲基-1,5-苯并二氮杂卓化合物为例,对制备的双酸催化剂的催化活性进行评价。取7个洁净的50ml圆底烧瓶,同时加入2mmol邻苯二胺,2mmol乙酰乙酸乙酯,2mmol苯乙醛和5ml无水乙醇,分别将0mg,5mg,10mg,15mg、20mg、25mg、30mg实施例1制得的cofe2o4@sio2@aptes@ata@cecl3分别加入上述7个圆底烧瓶中,将以上得到的混合物在室温下搅拌反应,通过tlc指示反应完成后,使用外部磁铁分离催化剂,并用无水乙醇和去离子水分别洗涤三次,在80℃下真空干燥24h,以便循环使用。圆底烧瓶中的反应溶液通过旋蒸,然后加入乙醇重结晶获得白色固体,抽滤后得到2-苄基-3酯基-4-甲基-1,5-苯并二氮杂卓化合物。具体反应方程式如下:催化剂活性评价结果如表1所示。表1结果证明,实施例1制备的磁性纳米双酸催化剂(cofe2o4@sio2@aptes@ata@cecl3)在一锅三组分串联催化合成1,5-苯并二氮杂卓类化合物的反应中展示了优异的催化性能,其最优的催化剂用量为20mg时,反应历经2h就能完成,目标产物的产率能达到90%,制备的产物的纯度可达96-98%。将上述反应体系中回收的催化剂重新按照上述方法进行催化合成2-苄基-3酯基-4-甲基-1,5-苯并二氮杂卓化合物的反应,催化剂用量为20mg,反应时间为2h,每次反应结束后外加磁铁分离催化剂,并用无水乙醇和去离子水洗涤3次,作为下一次催化反应的催化剂。重复使用6次,仍可以保持很高的催化活性,循环第6次的产率为85.9%。证明,本实施例制备的磁性纳米双酸催化剂催化合成1,5-苯并二氮杂卓类化合物的催化活性高,易回收且循环利用能力很好。将本发明实施例1中步骤a至步骤d的反应温度、反应时间、原料用量和反应溶剂,替换为本发明限定的其他条件,均可达到与实施例1相当的技术效果。按照与上述方法相同的方法对实施例2-4制备的磁性纳米双酸催化剂进行催化活性评价,催化剂的用量均为20mg,反应时间2h,其余反应条件完全相同。结果如表2所示。表2实施例2实施例3实施例4产率(%)838085以实施例2-4制备的双酸催化剂作为催化剂制备所得的2-苄基-3酯基-4-甲基-1,5-苯并二氮杂卓化合物的纯度为95-98%。循环使用6次后,产率仍可达到78-85%。以本发明实施例1制备的cofe2o4@sio2@aptes@ata@cecl3纳米双酸催化剂催化合成上述限定的其他1,5-苯并二氮杂卓类化合物,均可使最终产物的收率达到85-90%,纯度达到96-98%。以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换或改进等,均应包含在本发明的保护范围之内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1