一种用于强极性药物分离的硅胶基质色谱填料的制备方法
本发明属于新型色谱填料制备领域,尤其涉及一种用于强极性药物色谱分离的硅胶基质色谱填料的制备方法。
背景技术:
近期,随着生物医药行业的不断发展,强极性药物(人参皂苷类、核苷类、糖类氨基酸、生物碱类等)引起科研工作者的广泛关注。对强极性药物进行分离分析,对提高药物安全性及分析药物代谢过程等具有重要意义。
如今,在强极性药物的众多分析检测方法中,高效液相色谱法最为常用,然而,传统的反相色谱柱难以对其有效分离。因此,发展新型色谱分离材料具有重要的意义。目前,为了解决强极性药物的色谱分离难题,研究者发展了一系列新型色谱固定相,包括离子液体修饰多孔硅胶、碳点修饰多孔硅胶及聚合物修饰多孔硅胶等新型色谱分离材料,例如cn101757897a公开了一种壳寡糖亲水色谱固定相的制备方法,采用“链接化学”作为键合反应方法键合壳寡糖,首先在硅胶表面引入末端炔基,然后以水、甲醇或这些溶剂的混合体为反应溶剂,将修饰有叠氮基团的壳寡糖键合到硅胶表面,即得壳寡糖亲水色谱固定相。
然而,强极性药物仍难以有效分离。因此,发展选择性独特的新型液相色谱固定相,对解决上述问题具有重要意义。
技术实现要素:
针对现有技术存在的上述不足,本发明的目的在于提供一种用于强极性药物色谱分离的硅胶基质色谱填料的制备方法。
为实现以上目的,本发明提供的一种用于强极性药物色谱分离的硅胶基质色谱填料的制备方法,采用如下技术方案:
一种用于强极性药物分离的硅胶基质色谱填料的制备方法,包含以下步骤:
1)硅胶选择及活化处理:选取粒径2~5μm、孔径15~30nm的多孔硅胶微球作为载体,采用质量分数8~20%的稀硝酸对多孔硅胶微球进行浸泡处理后,采用去离子水洗涤至上清液呈中性,再采用无水乙醇洗涤1~3次,烘干;
2)硅胶表面修饰双键:以无水甲苯为反应介质,加入乙烯基或丙烯基硅烷化试剂,加热回流8~24h,依次采用无水甲苯和乙醇或甲醇进行洗涤,得到双键修饰的多孔硅胶微球,干燥;
3)预聚合体系构建:选取一种或两种及以上核苷类药物作为模板分子,以甲基丙烯酸或丙烯酰胺作为功能单体,以甲醇、乙醇或乙腈作为致孔剂,-5~10℃下预聚合5~20h,得到预聚合体系;
4)聚合反应:向预聚合体系中缓慢添加(液体约1~5秒一滴,固体约1~5秒10毫克)引发剂、交联剂和双键修饰的多孔硅胶微球,在30~70℃条件下反应12~48h;
5)模板分子洗脱:采用洗脱溶剂对色谱分离材料洗脱6~18次,烘干备用。
优选的,所述步骤1)中,采用真空烘箱进行烘干,所述真空烘箱温度设置为50~100℃,时间5~16h。
优选的,所述步骤2)中,采用所述甲苯洗涤2~5次,采用所述甲醇或乙醇洗涤2~5次。
优选的,所述步骤2)中,采用加热干燥,所述加热干燥温度为50~80℃,时间为6~16h。
优选的,所述步骤4)中,所述引发剂为偶氮二异丁腈(aibn);交联剂为三羟甲基丙烷三甲基丙烯酸酯(trim)、乙二醇二甲基丙烯酸酯(egdma)或二乙基苯(dvb)中的一种或两种以上。
优选的,步骤(5)所述的烘干温度为50~70℃,时间为8~24h。
所述步骤1)中稀硝酸活化可以有效减少硅胶材料表面的金属杂质,增加硅胶表面的硅羟基含量,利于后续的硅烷化修饰及印迹聚合反应在多孔硅胶表面进行。
所述步骤2)中,修饰乙烯基或丙烯基,一方面利于引导印迹聚合反应在多孔硅胶表面有序可控的进行,另一方面利于将硅胶表面的硅羟基进行封端处理。利于有效改善材料的色谱分离性能。
所述步骤3)中,核苷类药物可以通过市购得到,国产或进口核苷类药物均可,要满足三个条件:1)分子自身稳定性较好,不含有能够阻止或影响聚合反应的化学基团;2)分子具有羧基、羟基等可以与功能单体相互作用的基团;3)印迹过程中模板分子的刚性较强,且便于洗脱。
在本发明的一些优选实施方式中,所述核苷类药物为齐多夫定、拉米夫定或腺苷中的任意一种或几种的组合。
所述步骤3)中,以甲基丙烯酸或丙烯酰胺作为功能单体,甲基丙烯酸具有羧基,丙烯酰胺具有氨基,可分别与模板分子作用,形成分子印迹聚合物空腔,所形成的聚合物分别呈现不同的带电特性,可以实现不同的分离作用。
所述步骤5)中洗脱溶剂为甲醇、乙腈、乙酸、甲酸、水等溶剂中一种或两种及以上。
所述步骤1)中,采用质量分数8~20%的稀硝酸对多孔硅胶微球进行浸泡处理时间为1~5h。
本发明所述多孔硅胶微球可以通过市购得到,进口或国产多孔硅胶微球均可。
与现有技术相比,本发明以多孔硅胶微球为载体,选取核苷类药物为模板分子,借助表面印迹技术提供了一种新型硅胶基质色谱填料的制备方法,本发明将分子印迹聚合物优异的选择性与硅胶材料良好的孔结构、形貌特征、机械强度等性质相结合,通过控制反应过程中反应温度、反应时间、滴加试剂速度等,使所得色谱填料既具有较高的色谱分离柱效、较高的分离选择性、良好的色谱峰对称性,又具有良好的稳定性。
附图说明
通过阅读参照以下附图对非限制性实施例所作的详细描述,本发明的其它特征、目的和优点将会变得更明显:
图1为实施例1用于强极性药物分离的硅胶基质色谱填料的制备方法示意图;
图2为实施例1所用的多孔硅胶微球(裸硅胶载体)(a)与核苷类药物齐多夫定表面印迹色谱填料(b)的扫描电镜图;
图3为实施例1所用的多孔硅胶微球(a)、核苷类药物齐多夫定表面印迹色谱填料(b)与非印迹材料(c)的红外光谱图;
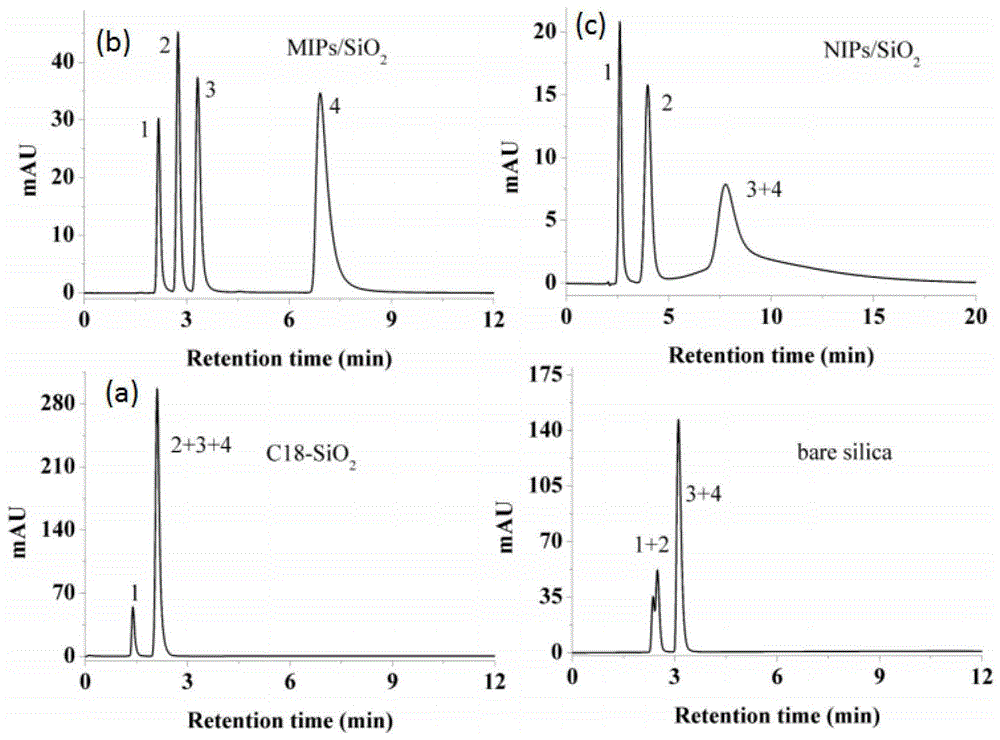
图4为c18填料(a)、核苷类药物齐多夫定表面印迹色谱填料(b)、非印迹材料对四种核苷类药物(c)的色谱分离图;
图5为核苷类药物齐多夫定表面印迹色谱填料对人参皂苷类化合物的色谱分离图。
图6为核苷类药物齐多夫定表面印迹色谱填料对核苷类药物连续分离结果重现性。
具体实施方式
下面结合具体实施例对本发明进行详细说明。以下实施例将有助于本领域的技术人员进一步理解本发明,但不以任何形式限制本发明。应当指出的是,对本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进。这些都属于本发明的保护范围。
实施例1
如图1所示,一种用于强极性药物分离的硅胶基质色谱填料的制备方法,包含以下步骤:
(1)硅胶选择及活化处理:选取粒径5μm,孔径20nm的多孔硅胶微球(购自日本fuji)作为载体,采用质量分数15%的稀硝酸对其进行浸泡处理2h后,采用去离子水洗涤至上清液呈中性,再采用无水乙醇洗涤3次,在55℃条件下8h真空烘干。
(2)硅胶表面修饰双键:以无水甲苯为反应介质,加入硅烷化试剂γ-甲基丙烯酰氧丙基三甲氧基硅烷(γ-mps,kh570),加热回流24h,依次采用无水甲苯洗涤1次、无水乙醇洗涤3次,得到双键修饰的多孔硅胶微球,在55℃条件下加热8h烘干。
(3)预聚合体系构建:选取齐多夫定作为模板分子,以甲基丙烯酸作为功能单体,以甲醇为致孔剂,在4℃条件下预聚合12h。
(4)聚合反应:向预聚合体系中缓慢添加引发剂偶氮二异丁腈(aibn)、交联剂乙二醇二甲基丙烯酸酯(egdma)及双键修饰(双键功能化)的多孔硅胶微球,反应条件50℃,反应时间12h,之后,继续升温至60℃,继续反应24h;
(5)模板分子洗脱:以比例为9:1的甲醇-甲酸混合溶液洗脱溶剂,洗涤材料6次,无水乙醇洗涤材料2次,55℃烘干8h备用。
由表征结果可知,所得材料粒径均匀分布在5μm,具有较好的球形形貌(见图2);红外表征结果证明分子印迹聚合物被成功修饰于多孔硅胶表面(见图3)。所得核苷类药物齐多夫定表面印迹色谱填料对四种核苷类药物(1-腺嘌呤、2-阿昔洛韦、3-司他夫定、4-齐多夫定)可实现基线分离,对四种目标物的分离柱效分别为15,300、19,300、17,100、12,600理论塔板数每米,对其分离结果优于c18以及非印迹材料的分离结果(见图4)。此外,所得印迹材料还可对人参皂苷类化合物实现基线分离,参见图5,其中对于人参皂苷rb1的分离柱效可达8,600理论塔板数每米;所得印迹材料连续使用17天后,柱效、保留因子、峰面积、色谱峰形对称性等良好,展现出很好的稳定性,参见图6。
实施例2
一种用于强极性药物分离的硅胶基质色谱填料的制备方法,包含以下步骤:
(1)硅胶选择及活化处理:选取粒径5μm,孔径15nm的多孔硅胶微球(购自日本fuji)作为载体,采用质量分数10%的稀硝酸对其进行浸泡处理1.5h后,采用去离子水洗涤至上清液呈中性,再采用无水乙醇洗涤2次,在60℃条件下8h真空烘干。
(2)硅胶表面修饰双键:以无水甲苯为反应介质,加入硅烷化试剂γ-甲基丙烯酰氧丙基三甲氧基硅烷(γ-mps),加热回流12h,依次采用无水甲苯洗涤2次、无水乙醇洗涤2次,得到双键修饰的多孔硅胶微球,在60℃条件下加热8h烘干。
(3)预聚合体系构建:选取齐多夫定和拉米夫定作为双模板分子,以丙烯酰胺作为功能单体,以乙腈为致孔剂,在8℃条件下预聚合16h。
(4)聚合反应:向预聚合体系中缓慢添加引发剂偶氮二异丁腈(aibn)、交联剂二乙基苯(dvb)、及双键功能化的多孔硅胶微球,反应条件58℃,反应时间15h,之后,继续升温至60℃,继续反应18h;(5)模板分子洗脱:以比例为8:3的乙醇-乙酸混合溶液洗脱溶剂,洗涤材料15次,无水乙醇洗涤材料1次,65℃烘干18h备用。
实施例3
一种用于强极性药物分离的硅胶基质色谱填料的制备方法,包含以下步骤:
(1)硅胶选择及活化处理:选取粒径3μm,孔径16nm的多孔硅胶微球(购自天津博瑞)作为载体,采用质量分数20%的稀硝酸对其进行浸泡处理1h后,采用去离子水洗涤至上清液呈中性,再采用无水乙醇洗涤1次,在100℃条件下5h真空烘干。
(2)硅胶表面修饰双键:以无水甲苯为反应介质,加入硅烷化试剂γ-甲基丙烯酰氧丙基三甲氧基硅烷(γ-mps),加热回流8h,依次采用无水甲苯洗涤5次、无水乙醇洗涤5次,得到双键修饰的多孔硅胶微球,在70℃条件下加热7h烘干。
(3)预聚合体系构建:选取齐多夫定和腺苷作为双模板分子,以丙烯酰胺作为功能单体,以乙腈为致孔剂,在8℃条件下预聚合16h。
(4)聚合反应:向预聚合体系中缓慢添加引发剂偶氮二异丁腈(aibn)、交联剂三羟甲基丙烷三甲基丙烯酸酯(trim)、及双键功能化的多孔硅胶微球,反应条件60℃,反应时间12h,之后,继续升温至70℃,继续反应12h;
(5)模板分子洗脱:以比例为6:1的乙腈-乙酸混合溶液洗脱溶剂,洗涤材料10次,无水乙醇洗涤材料2次,70℃烘干15h备用。
实施例4
一种用于强极性药物分离的硅胶基质色谱填料的制备方法,包含以下步骤:
(1)硅胶选择及活化处理:选取粒径5μm,孔径30nm的多孔硅胶微球(购自日本富士)作为载体,采用质量分数15%的稀硝酸对其进行浸泡处理3h后,采用去离子水洗涤至上清液呈中性,再采用无水乙醇洗涤1次,在100℃条件下5h真空烘干。
(2)硅胶表面修饰双键:以无水甲苯为反应介质,加入硅烷化试剂γ-甲基丙烯酰氧丙基三甲氧基硅烷(γ-mps),加热回流8h,依次采用无水甲苯洗涤5次、无水乙醇洗涤5次,得到双键修饰的多孔硅胶微球,在70℃条件下加热7h烘干。
(3)预聚合体系构建:选取齐多夫定和腺苷作为双模板分子,以丙烯酰胺作为功能单体,以乙腈为致孔剂,在-5℃条件下预聚合16h。
(4)聚合反应:向预聚合体系中缓慢添加引发剂偶氮二异丁腈(aibn)、交联剂三羟甲基丙烷三甲基丙烯酸酯(trim)、及双键功能化的多孔硅胶微球,反应条件30℃,反应时间12h,之后,继续升温至65℃,继续反应12h;
(5)模板分子洗脱:以比例为6:1的乙腈-乙酸混合溶液洗脱溶剂,洗涤材料10次,无水乙醇洗涤材料2次,70℃烘干15h备用。
以上对本发明的具体实施例进行了描述。需要理解的是,本发明并不局限于上述特定实施方式,本领域技术人员可以在权利要求的范围内做出各种变形或修改,这并不影响本发明的实质内容。
- 还没有人留言评论。精彩留言会获得点赞!