一种应用于催化合成生物柴油的固体酸材料及制备方法
本发明属于物理化学和材料化学的范畴,公开了一种应用于催化合成生物柴油的固体酸材料及制备方法。
背景技术:
进入21世纪以来,能源危机和环境污染已经成为全人类面临的重大课题。石油供需矛盾日益突出,研究新的可替代能源成为当务之急。尤其对于我国而言,近年随着经济飞速发展石油需求量急剧增长,供应缺口越来越大。在这种背景下各国纷纷加快石化燃料替代能源开发的步伐,其中生物柴油以其优越的环保性能受到了各国的重视。生物柴油的主要成分是不同链长的脂肪酸甲酯,一般通过动植物油脂和甲醇进行酯交换反应制备。生物柴油的生产方法可根据所使用催化剂的种类,分为酸催化和碱催化。虽然碱性催化剂能够在温和条件下催化酯交换反应,但容易引发油脂皂化,并且碱性催化剂易受到油脂中水和游离脂肪酸的影响而失活。酸性催化剂不仅能够催化酸价高的油脂,而且可以催化酯交换反应和酯化反应的同时进行,从而提高生物柴油的产率。因此,酸催化酯交换制备生物柴油的应用范围更广。氧化锆作为一种超强酸催化剂,可以高效催化酯化反应及酯交换反应。
kiss等研究表明,硫酸化氧化锆催化醇和游离脂肪酸酯化反应中具有很好的活性、选择性。这为硫酸化氧化锆作为固体酸催化剂制备生物柴油打下了理论基础。garcia利用无溶剂方法制备的硫酸化氧化锆在120℃下能获得100%的大豆油转化率,但是快速失活和硫元素的流失等不足严重的限制了其应用。sivakumar在合成硫酸化氧化锆的过程中引入了铁和锰等元素。其合成的材料在动物脂肪与甲醇的酯交换催化过程中,催化5次后生物柴油产率仍在90%以上,展现出良好的催化活性及稳定性。
技术实现要素:
针对上述情况,本发明的目的在于:提供一种应用于催化合成生物柴油的固体酸材料及制备方法,其能够实现高转化率、高选择性的催化大豆油与甲醇酯交换制备生物柴油。
为实现上述目的,本发明所采取的技术方案是:提供一种应用于催化合成生物柴油的固体酸材料,其具体是以锆源、锡源、有机胺、有机铵盐、氨水、硫酸、无水乙醇和去离子水为原料,原料用量满足锆源∶有机胺:有机铵盐∶锡源:无水乙醇=900~1820∶125~270∶250~400∶25~140∶100000的摩尔配料比。
本发明所述的固体酸催化材料中,所述原料锆源为四水硫酸锆、正丁醇锆、五水硝酸锆、八水氯氧化锆中的任意一种或几种的混合物。
所述锡源,选用四溴化锡、无水氯化锡、五水氯化锡中的任意一种。
所述有机胺,选用三乙醇胺或三异丙醇胺中的任意一种。
所述有机铵盐,选用十六烷基三甲基氯化铵、双辛烷基二甲基氯化铵、双辛烷基二甲基溴化铵、双癸烷基二甲基氯化铵或双癸烷基二甲基溴化铵中的任意一种。
本发明所述的固体酸催化材料具有纳米堆积介孔结构、较大的比表面积和孔体积,介孔孔径可调,其介孔孔径2~30nm,比表面积110~190m2/g,孔体积0.16~0.3cm3/g范围内可调。
进而,本发明所述的一种应用于催化合成生物柴油的固体酸材料的制备方法,具体是按照下述方法制备得到的:
(1)按照锆源∶有机胺:有机铵盐∶锡源:无水乙醇=900~1820∶125~270∶250~400∶25~140∶100000的摩尔配料比,将锆源、锡源、有机胺、有机铵盐依次完全溶解到无水乙醇中得到澄清的溶液;
(2)将所述澄清溶液于水热釜中60~90℃下,预晶化处理0.5~3h,得到预晶化产物;
(3)向预晶化产物中滴加氨水调节其ph=9;
(4)处理过后的产物于水热釜中100~140℃再次晶化,24~48h;
(5)使用无水乙醇洗涤二次晶化后的产物至中性,将洗涤后的产物于60~80℃干燥;
(6)干燥所得产物在300~350℃下焙烧,脱除有机模板剂;
(7)脱除有机模板剂的每1g产物以10~20ml浓度为0.5~2mol/l的硫酸溶液浸渍,浸渍时间为0.5~3h;
(8)浸渍完成后的溶液经过抽滤后,将滤饼在100~120℃下干燥;
(9)干燥后的产物在450~600℃下焙烧3~5h得到so42-/sno2-zro2固体酸催化材料。
本发明所述给出的so42-/sno2-zro2固体酸催化材料的制备方法在乙醇作为溶剂的合成体系下,有机铵盐在乙醇中形成互相交联的网状结构胶束,并与有机胺螯合的锆源、锡源形成团聚的前驱体溶液。在一定温度下预晶化处理,这个过程中部分的螯合锆源、锡源通过缓慢的水解形成相应的羟基化合物。在预晶化结束后,通过滴加氨水引入更多的羟基基团,同时通过控制滴加氨水的速率来达到水解和团聚的平衡。在碱性溶液中,锆源逐渐的水解和聚合形成有机铵盐模板剂与锆、锡水解产物的结合体。通过高温焙烧过程脱出有机模板剂,在模板剂存在的位置形成颗粒之间的介孔孔道。在硫酸浸渍过程中,硫酸吸附于sno2-zro2的表面。通过进一步的焙烧过程,制备得到具有较高比表面积、孔径和孔体积较大、锡与硫高度分散的so42-/sno2-zro2固体酸催化材料。在此过程中,通过调控有机铵盐离子表面活性剂与有机胺的引入种类、晶化温度、焙烧温度及锡源、硫源的引入量,调控所得材料的比表面积、孔体积、介孔孔径;通过优化焙烧条件(包括温度和升温速率),调节氧化锆的晶化程度,进而调控所得材料氧化锆的晶相结构、晶粒尺寸;通过调控锡、硫的加入量调控其在氧化锆中的引入量。
本发明的有益效果是:所给出的so42-/sno2-zro2固体酸催化材料可以作为催化剂,用于催化大豆油与甲醇酯交换制备生物柴油;其可显示出极高的大豆油与甲醇酯交换活性、生物柴油选择性。在140℃、甲醇与三油酸甘油酯摩尔比为20:1、反应5h、催化剂用量为大豆油质量的3%条件下,大豆油转化率不低于99%,产物中的生物柴油含量不低于97%;此外,本发明制备的so42-/sno2-zro2固体酸催化材料具有优异的在生性能,在重复使用7次后,其结构、织构及大豆油与甲醇酯交换反应催化性能未发生变化;制备方法过程简单易行,重现率高。
附图说明
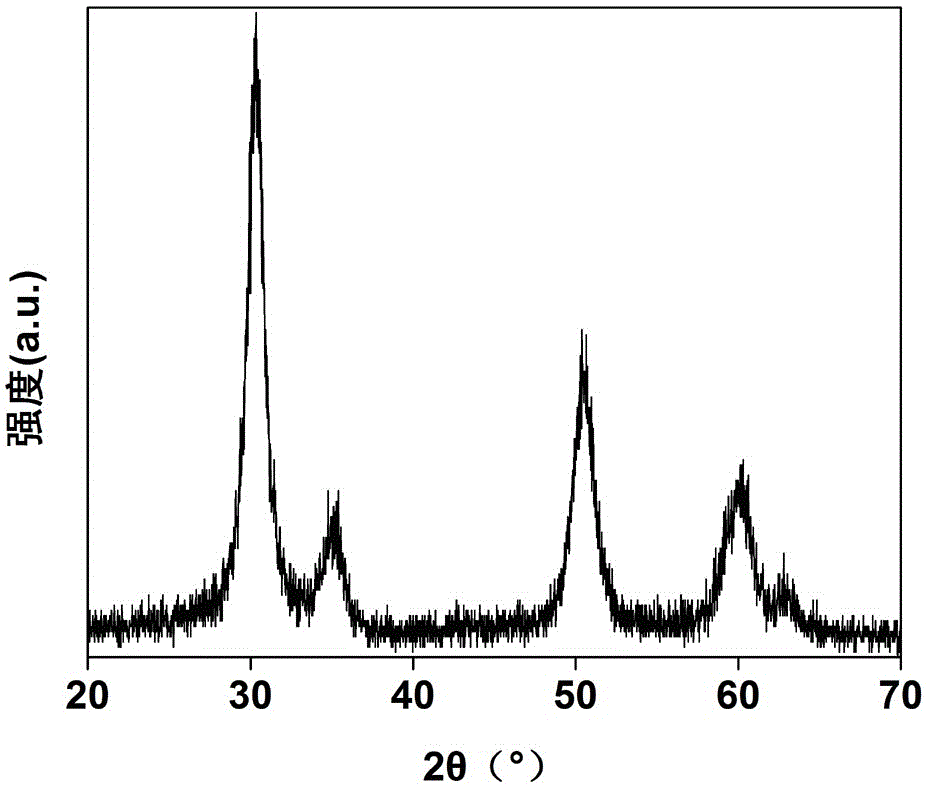
图1是so42-/sno2-zro2固体酸催化材料的xrd谱图;
图2是so42-/sno2-zro2固体酸催化材料的sem照片;
图3是so42-/sno2-zro2固体酸催化材料的高分辨tem照片;
图4是so42-/sno2-zro2固体酸催化材料的n2吸附-脱附等温线;
图5是so42-/sno2-zro2固体酸催化材料的n2孔径分布曲线;
图6是so42-/sno2-zro2固体酸催化材料的nh3-tpd图谱;
图7是so42-/sno2-zro2固体酸催化材料在催化大豆油与甲醇反应中不同反应时间的大豆油转化率及生物柴油选择性;
图8是so42-/sno2—zro2固体酸催化材料催化性能随催化反应次数变化的趋势图。
具体实施方式
下面结合实施例对本发明的具体实施方式作进一步描述。以下实施例仅用于更加清楚地说明本发明的技术方案,而不是限制本发明的保护范围。本领域普通技术人员在不脱离本发明原理和宗旨的情况下,针对这些实施例进行的各种变化、修改、替换和变型,均应包含在本发明的保护范围之内。
实施例1
一种应用于催化合成生物柴油的固体酸材料的制备方法,具体制备制备过程如下:(1)在32℃强烈搅拌下,依次将4g硝酸锆五水合物、0.2g五水四氯化锡、0.3g三乙醇胺溶解于50ml无水乙醇中得到澄清溶液,计为溶液a;将0.8g双八烷基二甲基氯化铵溶解于10ml无水乙醇中,计为溶液b;将双八烷基二甲基氯化铵乙醇溶液缓慢的加入到溶液a中;
(2)将混合后的溶液转移到带有100ml聚四氟乙烯内衬的水热釜中,在70℃处理4h,得到预晶化产物;
(3)预晶化产物转移至玻璃烧杯中,在搅拌条件下,缓慢滴加氨水调节溶液ph=9;
(4)将上述产物转移到带有100ml聚四氟乙烯内衬的水热釜中,在100℃处理48h,得到二次晶化产物;
(5)使用无水乙醇将二次晶化产物洗涤至中性,在60℃下干燥24h;
(6)将干燥后的样品以1℃/min的升温速率升温至300℃焙烧5h,脱除存在于介孔孔道内的有机模板剂;
(7)每1g脱除模板剂的样品使用10ml浓度为1.5mol/l的硫酸浸渍0.5h;
(8)浸渍完成后,通过减压抽滤将多余的硫酸溶液滤出,所得滤饼在100℃下干燥18h;
(9)将干燥后的浸渍样品以1℃/min的升温速率升温至550℃焙烧3h,制得so42-/sno2-zro2固体酸催化材料。
实施例2
一种应用于催化合成生物柴油的固体酸材料的制备方法,具体制备制备过程如下:
(1)32℃强烈搅拌下,依次将6g八水合氯化锆、0.07g无水四氯化锡、0.25g三异丙醇胺溶解于50ml无水乙醇中得到澄清溶液,计为溶液a;将1.1g双癸烷基二甲基氯化铵溶解于10ml无水乙醇中,计为溶液b;将双癸烷基二甲基氯化铵乙醇溶液缓慢的加入到溶液a中;
(2)将混合后的溶液转移到带有100ml聚四氟乙烯内衬的水热釜中,在80℃处理4h,得到预晶化产物;
(3)将预晶化产物转移至玻璃烧杯中,在搅拌条件下,缓慢滴加氨水调节溶液ph=9;
(4)将上述产物转移到带有100ml聚四氟乙烯内衬的水热釜中,在120℃处理24h,得到二次晶化产物;
(5)使用无水乙醇将二次晶化产物洗涤至中性,在70℃下干燥12h;
(6)将干燥后的样品以1℃/min的升温速率升温至300℃焙烧5h,脱除存在于介孔孔道内的有机模板剂;
(7)脱除有机模板剂的每1g样品使用15ml浓度为1mol/l的硫酸浸渍1h;
(8)浸渍完成后,通过减压抽滤将多余的硫酸溶液滤出,所得滤饼在110℃下干燥12h;
(9)将干燥后的浸渍样品以1℃/min的升温速率升温至300℃焙烧5h,制得so42-/sno2-zro2固体酸催化材料。
实施例3
一种应用于催化合成生物柴油的固体酸材料的制备方法,具体制备制备过程如下:
(1)32℃强烈搅拌下,依次将5g四水硫酸锆、0.11g四溴化锡、0.3g三异丙醇胺溶解于50ml无水乙醇中得到澄清溶液,计为溶液a;将1.2g十六烷基三甲基氯化铵溶解于10ml无水乙醇中,计为溶液b;将十六烷基三甲基氯化铵乙醇溶液缓慢的加入到溶液a中;
(2)将混合后的溶液转移到带有100ml聚四氟乙烯内衬的水热釜中,在90℃处理3h,得到预晶化产物;
(3)将预晶化产物转移至玻璃烧杯中,在搅拌条件下,缓慢滴加氨水调节溶液ph=9;
(4)将上述产物转移到带有100ml聚四氟乙烯内衬的水热釜中,在140℃处理24h,得到二次晶化产物;
(5)使用无水乙醇将二次晶化产物洗涤至中性,在80℃下干燥12h;
(6)将干燥后的样品以1℃/min的升温速率升温至300℃焙烧5h,脱除存在于介孔孔道内的有机模板剂;
(7)脱除有机模板剂的每1g样品使用20ml浓度为0.5mol/l的硫酸浸渍3h;
(8)浸渍完成后,通过减压抽滤将多余的硫酸溶液滤出,所得滤饼在120℃下干燥6h;
(9)将干燥后的浸渍样品以1℃/min的升温速率升温至500℃焙烧4h,制得so42-/sno2-zro2固体酸催化材料。
实施例4
一种应用于催化合成生物柴油的固体酸材料的制备方法,具体制备制备过程如下:
(1)32℃强烈搅拌下,依次将6g硝酸锆五水合物、0.5g五水四氯化锡、0.2g三乙醇胺溶解于50ml无水乙醇中得到澄清溶液,计为溶液a;将1g十六烷基三甲基氯化铵溶解于10ml无水乙醇中,计为溶液b;将十六烷基三甲基氯化铵乙醇溶液缓慢的加入到溶液a中;
(2)将混合后的溶液转移到带有100ml聚四氟乙烯内衬的水热釜中,在80℃处理4h,得到预晶化产物;
(3)将预晶化产物转移至玻璃烧杯中,在搅拌条件下,缓慢滴加氨水调节溶液ph=9;
(4)将上述产物转移到带有100ml聚四氟乙烯内衬的水热釜中,在120℃处理36h,得到二次晶化产物;
(5)使用无水乙醇将二次晶化产物洗涤至中性,在80℃下干燥24h;
(6)将干燥后的样品以1℃/min的升温速率升温至300℃焙烧6h,脱除存在于介孔孔道内的有机模板剂;
(7)脱除有机模板剂的每1g样品使用10ml浓度为2mol/l的硫酸浸渍2h;
(8)浸渍完成后,通过减压抽滤将多余的硫酸溶液滤出,所得滤饼在100℃下干燥12h;
(9)将干燥后的浸渍样品以1℃/min的升温速率升温至450℃焙烧5h,制得so42-/sno2-zro2固体酸催化材料。
实施例5
一种应用于催化合成生物柴油的固体酸材料的制备方法,具体制备制备过程如下:
(1)32℃强烈搅拌下,依次将5g八水合氯氧化锆、0.21g无水四氯化锡、0.4g三乙醇胺溶解于50ml无水乙醇中得到澄清溶液,计为溶液a;将1.25g双八烷基二甲基溴化铵溶解于10ml无水乙醇中,计为溶液b;将双八烷基二甲基溴化铵乙醇溶液缓慢的加入到溶液a中;
(2)将混合后的溶液转移到带有100ml聚四氟乙烯内衬的水热釜中,在80℃处理4h,得到预晶化产物;
(3)将预晶化产物转移至玻璃烧杯中,在搅拌条件下,缓慢滴加氨水调节溶液ph=9;
(4)将上述产物转移到带有100ml聚四氟乙烯内衬的水热釜中,在120℃处理24h,得到二次晶化产物;
(5)使用无水乙醇将二次晶化产物洗涤至中性,在60℃下干燥12h;
(6)将干燥后的样品以1℃/min的升温速率升温至350℃焙烧3h,脱除存在于介孔孔道内的有机模板剂;
(7)脱除有机模板剂的每1g样品使用15ml浓度为1mol/l的硫酸浸渍3h;
(8)浸渍完成后,通过减压抽滤将多余的硫酸溶液滤出,所得滤饼在120℃下干燥6h;
(9)将干燥后的浸渍样品以1℃/min的升温速率升温至500℃焙烧4h,制得so42-/sno2-zro2固体酸催化材料。
实施例6
一种应用于催化合成生物柴油的固体酸材料的制备方法,具体制备制备过程如下:
(1)32℃强烈搅拌下,依次将4g正丁醇锆、0.36g四溴化锡、0.4g三异丙醇胺溶解于50ml无水乙醇中得到澄清溶液,计为溶液a;将1.25g双癸烷基二甲基溴化铵溶解于10ml无水乙醇中,计为溶液b;将双癸烷基二甲基溴化铵乙醇溶液缓慢的加入到溶液a中;
(2)将混合后的溶液转移到带有100ml聚四氟乙烯内衬的水热釜中,在70℃处理4h,得到预晶化产物;
(3)将预晶化产物转移至玻璃烧杯中,在搅拌条件下,缓慢滴加氨水调节溶液ph=9;
(4)将上述产物转移到带有100ml聚四氟乙烯内衬的水热釜中,在140℃处理24h,得到二次晶化产物;
(5)使用无水乙醇将二次晶化产物洗涤至中性,在60℃下干燥12h;
(6)将干燥后的样品以1℃/min的升温速率升温至330℃焙烧5h,脱除存在于介孔孔道内的有机模板剂;
(7)脱除有机模板剂的每1g样品使用20ml浓度为0.5mol/l的硫酸浸渍0.5h;
(8)浸渍完成后,通过减压抽滤将多余的硫酸溶液滤出,所得滤饼在100℃下干燥12h;
(9)将干燥后的浸渍样品以1℃/min的升温速率升温至550℃焙烧5h,制得so42-/sno2-zro2固体酸催化材料。
如图1所示,为so42-/sno2-zro2固体酸催化材料的xrd谱图,可以看出,样品在2θ=30.2°,35.3°,50.3°,60.2°和62.8°具有明显的衍射峰,证明氧化锆为四方相结构。同时没有发现氧化锡的特征衍射峰,说明氧化锡均匀的分散与氧化锆中;此外通过谢乐公式对氧化锆晶粒尺寸计算可得其尺寸为10nm左右。
如图2、3所示,是so42-/sno2-zro2固体酸催化材料的sem照片及高分辨tem照片可以看出材料呈现纳米堆积结构,从高分辨tem照片(b)可以看出材料晶粒尺寸为10nm左右,与上述xrd计算结果相一致。
从图4材料的n2吸附-脱附等温线可见,样品呈现典型的ⅳ型吸附等温线及h3型滞后环,说明固体材料含有颗粒堆积的不规则孔道。
从图5孔径分布曲线可见,样品的孔道分布较宽。样品的比表面积和孔体积分别为190m2/g和0.3cm3/g,平均介孔孔径为4.9nm。
如图6所示,可以看出通过高斯拟合可以看出在200~300℃的氨气脱附峰对应材料中的弱酸酸性位点,300~450℃的氨气脱附峰对应材料中的中强酸酸性位点,450~600℃的氨气脱附峰对应材料中的强酸酸性位点。从中可以看出材料主要的酸性位点为中强酸。
以上制备的样品作为大豆油与甲醇酯交换制备生物柴油的催化剂,考察催化剂的催化性能。反应在高压反应釜中进行,反应釜内催化剂为0.3g、大豆油为10g、甲醇为7.2g、反应温度为140℃,搅拌速率为600r/min,反应时间为分别为1h、2h、3h、4h、5h。反应产物在蒸馏出多余未反应的甲醇后,将产物上清液,在配有氢火焰离子化检测器(fid)的气相色谱仪上进行分析。催化评价结果如图7所示,催化材料在催化大豆油与甲醇反应中大豆油转化率与生物柴油选择性随反应时间的增长而增长,反应时间达5h时其大豆油转化率在99%以上、生物柴油选择性在97%以上,表明该催化材料具有优异的大豆油与甲醇酯交换反应催化活性与生物柴油选择性。此外,如图8所示,该催化材料在经过再生后重复使用的过程中,对大豆油的转化率和生物柴油的选择性未发生明显变化,表明该样品具有非常优异的重复使用性能。
综合上述实施例中所制得的产品,本发明所述的so42-/sno2-zro2固体酸催化材料具有纳米堆积介孔结构、较大的比表面积和孔体积,介孔孔径可调,其介孔孔径2~30nm,比表面积110~190m2/g,孔体积0.16~0.3cm3/g范围内可调。上述技术方案所给出的so42-/sno2-zro2固体酸催化材料可以作为催化剂,用于催化大豆油与甲醇酯交换制备生物柴油;其可显示出极高的大豆油与甲醇酯交换活性、生物柴油选择性。在140℃、甲醇与三油酸甘油酯摩尔比为20:1、反应5h、催化剂用量为大豆油质量的3%条件下,大豆油转化率不低于99%,产物中的生物柴油含量不低于97%;此外,本发明制备的so42-/sno2-zro2固体酸催化材料具有优异的在生性能,在重复使用7次后,其结构、织构及大豆油与甲醇酯交换反应催化性能未发生变化;制备方法过程简单易行,重现率高。
- 还没有人留言评论。精彩留言会获得点赞!