稀土氧化物催化剂及其制备方法和应用与流程
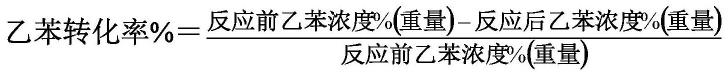
1.本发明涉及一种稀土氧化物催化剂以及稀土氧化物催化剂的制备方法及应用。
背景技术:
2.稀土氧化物常作为电子助剂和结构助剂用于修饰催化剂,从而提高催化剂的活性、选择性和稳定性。以ceo2作为助剂或者载体的催化剂在工业上应用很广泛,包括用于烷基芳烃脱氢、催化燃烧、氧化还原、催化裂解及三效催化等方面。铈的外层价电子结构为4f15d16s2,特殊的电子结构决定了它的可变价性,它有ceo2和ce2o3两种氧化物形态,因此具有良好的氧化还原性;而且可以快速达到ce
4+
/ce
3+
的平衡,具有极好的储氧效应和释放氧的能力。目前烷基芳烃脱氢催化剂大都为fe-k-ce-mo系催化剂,其中催化剂中稀土ce起到了极其重要的作用。
3.wo2008090974(用于烷基芳烃脱氢的高强度催化剂及其制备方法及应用)通过使用0.5~5微米的氢氧化碳酸铈作为原料,提高铈的含量可制备的催化剂可以有效提高催化剂的机械强度,用于烷基芳烃脱氢的工业生产中。
4.烷基芳烃脱氢生产烷烯基芳烃工艺,包括乙苯脱氢制苯乙烯,甲乙苯脱氢制甲基苯乙烯,二乙苯脱氢制二乙烯基苯等。以苯乙烯为例,目前全球的产能超过3500万吨/年,催化剂的年消耗量在15000吨以上,消耗的稀土量(以稀土氧化物计)超过1500吨,稀土耗量大。因此以高效利用稀土资源为目标,围绕高能耗化工过程的节能需求,从而达到降低苯乙烯生产成本、节能降耗、更高效生产苯乙烯的目的具有重要的意义。此外,稀土在电子、石油化工、环境保护、航天等多个领域都有着广泛的应用,尤其是在高新技术和国防工业中的不可替代性,是非再生性资源,从保护稀土资源的角度,高效利用稀土资源,也有着非常重要的意义。
技术实现要素:
5.本发明所要解决的技术问题之一是针对现有技术中烷基芳烃脱氢生产烷烯基芳烃反应存在副产物苯含量高的技术问题,提供一种稳定性高、催化剂的稀土元素用量少且分布均匀、降低副产物苯含量的稀土氧化物催化剂。
6.根据本发明的第一方面,本发明提供一种稀土氧化物催化剂,该催化剂包括稀土、铁、钾和可选地其他金属助剂组分,稀土含有ce和r,r为sc、y和la中的至少一种,(ce/r)
icp
=10~90,ce
eds
/ce
icp
=1.0~1.4,其中,(ce/r)
icp
是以等离子体原子发射光谱法测定的催化剂中稀土ce和r组分以元素计的重量比,ce
eds
/ce
icp
是以x射线能量谱仪表征的催化剂表面中稀土ce组分与以等离子体原子发射光谱法测定的催化剂中稀土ce组分以元素计的重量比。
7.优选地,所述催化剂中(ce/r)
icp
为20~50,ce
eds
/ce
icp
为1.1~1.3。
8.优选地,以元素计并以催化剂的总重量为基准,稀土组分的总含量为4~9%。
9.优选地,以元素计并以催化剂的总重量为基准,稀土r组分的含量为0.05~1%;稀
土ce组分的含量为3.8~8%。
10.优选地,铁组分的加入以元素计并以催化剂的总重量为基准,铁组分的含量为45~55%。
11.优选地,钾组分的加入以元素计并以催化剂的总重量为基准,钾组分的含量为7~13%。
12.优选地,其他金属助剂包括碱土金属、第ivb族金属、第vib族金属和第viib族金属中的至少一种,以元素计并以催化剂的总重量为基准,其他金属助剂含量为0.5-10%;更优选地,其他金属助剂包括碱土金属、第vib金属、第ivb族金属、第vib族金属和第viib族金属,以元素计并以催化剂的总重量为基准,碱土金属组分的含量为0.3~2.8%,第ivb族组分的含量为0.01~0.2%,第vib族组分的含量为0.3~2.5%,第viib族组分的含量为0.01~2.0%,优选以元素计并以催化剂的总重量为基准,所述其他金属助剂包括ca 0.3~2.8%,ti 0.01~0.2%,mo 0.3~2.5%,mn 0.01~2.0%。
13.根据本发明的第二方面,本发明提供一种稀土氧化物催化剂的制备方法,该方法包括以下步骤:
14.1)在含水溶剂存在下,将稀土组分r化合物、稀土组分铈化合物在碱性条件下接触,得到催化剂前体;
15.2)将铁源、钾源、可选地其他金属助剂源与粘结剂源混合均匀,然后加入所述催化剂前体,进行混捏、成型、干燥、焙烧。
16.优选地,所述制备方法包括:
17.1)将稀土组分r化合物用水作为溶剂形成溶液,然后加入碱性物质混匀,之后加稀土组分铈化合物,经静置得催化剂前体;
18.2)将铁源、钾源、可选地其他金属助剂源与粘结剂源混合均匀,然后加入所述催化剂前体,经湿捏、挤条、成型、干燥、焙烧后得所需的成品催化剂。
19.优选地,于80℃至380℃以下,优选340-380℃下焙烧4~10小时,然后于高于380℃至650℃以下,优选580-650℃下焙烧3~7小时,然后于高于650℃至900℃以下,优选800-880℃焙烧3~6小时。
20.优选地,步骤2)干燥的条件包括:室温~80℃,优选50-70℃,更优选干燥时间为3-5h。
21.优选地,所述步骤1)中水的用量为步骤1)和步骤2)原料总重量的15-30重量%,优选为步骤1)和步骤2)原料总重量的20-28重量%。
22.优选地,所述步骤1)接触的时间为4-24h。
23.优选地,稀土组分r化合物为r盐、r氢氧化物、r氧化物中的一种或多种,进一步优选为可溶性r盐;更优选为硝酸盐。
24.优选地,硝酸铈和草酸铈的混合物,以ceo2计,硝酸铈与草酸铈的重量比为0.3-2∶1。
25.优选地,形成碱性条件的碱性物质为尿素和/或氨。
26.优选地,形成碱性条件的碱性物质的用量为步骤1)和步骤2)原料总重量的1-3重量%。
27.优选地,稀土组分铈化合物为铈盐、铈氢氧化物和铈氧化物中的一种或多种,更优
选为硝酸铈、草酸铈和碳酸铈中的一种或多种。
28.根据本发明的第三方面,本发明提供一种所述的制备方法制备得到的稀土氧化物催化剂。
29.根据本发明的第四方面,本发明提供一种所述的稀土氧化物催化剂在制备烷烯基芳烃中的应用,优选所述烷烯基芳烃为苯乙烯、甲基苯乙烯和二乙烯基苯中的一种或多种。
30.与现有技术相比,本发明提供的稀土氧化物催化剂,采用微量的r元素,r为sc、y和la中的至少一种,降低了稀土元素的总量,保护稀土资源,高效利用稀土,且催化剂表面的稀土ce分布更多,更均匀,催化剂具有稳定性的特点。
31.本发明提供的稀土氧化物催化剂的制备方法,制备步骤中首先控制稀土ce与r在碱性条件下的接触,优选地再与铁钾混合分步焙烧,可以使ce富集高效分布在催化剂的表面。
32.本发明提供的催化剂应用于烷基芳烃脱氢生产烷烯基芳烃反应中能够明显降低副产物苯的含量。
具体实施方式
33.在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
34.本发明提供一种稀土氧化物催化剂,该催化剂包括稀土、铁、钾和可选地其他金属助剂组分,稀土含有ce和r,r为sc、y和la中的至少一种,(ce/r)
icp
=10~90,ce
eds
/ce
icp
=1.0~1.4,其中,(ce/r)
icp
是以等离子体原子发射光谱法测定的催化剂中稀土ce和r组分以元素计的重量比,ce
eds
/ce
icp
是以x射线能量谱仪表征的催化剂表面中稀土ce组分与以等离子体原子发射光谱法测定的催化剂中稀土ce组分以元素计的重量比;本发明通过在催化剂中添加微量的稀土r组分,使得本发明的催化剂能够降低稀土ce组分的使用量,降低稀土使用总量,高效利用稀土,保护稀土资源;同时微量的稀土r组分使稀土ce组分高效富集在催化剂的表面分布,催化剂应用于烷基芳烃脱氢生产烷烯基芳烃反应中能够降低副产物苯的含量,取得较好的技术效果。
35.根据本发明,优选地,所述催化剂中(ce/r)
icp
为20~50,ce
eds
/ce
icp
为1.1~1.3,使稀土ce组分富集高效分布在催化剂的表面,有利于在烷基芳烃脱氢生产烷烯基芳烃反应中降低副产物苯的含量。
36.根据本发明,优选地,以元素计并以催化剂的总重量为基准,稀土组分的总含量为4~9%,而现有技术稀土组分的含量通常高于10重量%,采用前述含量的稀土组分,催化剂能够实现在烷基芳烃脱氢生产烷烯基芳烃反应中降低副产物苯的含量,高效利用稀土,保护稀土资源。
37.根据本发明的一种优选实施方式,以元素计并以催化剂的总重量为基准,稀土r组分的含量为0.05~1%;稀土ce组分的含量为3.8~8重量%;本发明采用微量的稀土r组分,降低稀土ce的使用量,降低稀土使用总量,使稀土ce组分在催化剂表面均匀分布。
38.根据本发明的一种优选的实施方式,铁组分的加入以元素计并以催化剂的总重量
为基准,铁组分的含量为45~55%。
39.根据本发明的一种优选的实施方式,钾组分的加入以元素计并以催化剂的总重量为基准,钾组分的含量为7~13%。
40.根据本发明的一种优选实施方式,其他金属助剂包括碱土金属、第ivb族金属、第vib族金属和第viib族金属中的至少一种,以元素计并以催化剂的总重量为基准,其他金属助剂含量为0.5-10%;更优选地,其他金属助剂包括碱土金属、第vib金属、第ivb族金属、第vib族金属和第viib族金属,以元素计并以催化剂的总重量为基准,碱土金属组分的含量为0.3~2.8%,第ivb族组分的含量为0.01~0.2%,第vib族组分的含量为0.3~2.5%,第viib族组分的含量为0.01~2.0%。
41.根据本发明,优选以元素计并以催化剂的总重量为基准,所述其他金属助剂包括ca 0.3~2.8%,ti 0.01~0.2%,mo 0.3~2.5%,mn 0.01~2.0%。
42.本发明的催化剂具有前述组成和性质即可实现本发明的目的,对其制备方法无特殊要求,针对本发明,提供一种稀土氧化物催化剂的制备方法,该方法包括以下步骤:
43.1)在含水溶剂存在下,将稀土组分r化合物、稀土组分铈化合物在碱性条件下接触,得到催化剂前体;
44.2)将铁源、钾源、可选地其他金属助剂源与粘结剂源混合均匀,然后加入所述催化剂前体,进行混捏、成型、干燥、焙烧。
45.本发明提供的制备方法,控制稀土ce与r在碱性条件下的接触,再与铁钾混合接触,可以使稀土ce富集高效分布在催化剂的表面,有利于在烷基芳烃脱氢生产烷烯基芳烃反应中降低副产物苯的含量。
46.根据本发明的一种优选实施方式,所述制备方法包括:
47.1)将稀土组分r化合物用水作为溶剂形成溶液,然后加入碱性物质混匀,之后加稀土组分铈化合物,经静置得催化剂前体;
48.2)将铁源、钾源、可选地其他金属助剂源与粘结剂源混合均匀,然后加入所述催化剂前体,经湿捏、挤条、成型、干燥、焙烧后得所需的成品催化剂;采用前述制备方法,使稀土ce富集高效分布在催化剂的表面,有利于在烷基芳烃脱氢生产烷烯基芳烃反应中降低副产物苯的含量。
49.根据本发明的一种实施方式,所述步骤1)中的用于溶解稀土组分钇的水以催化剂的总重量为基准,水用量为15~30%。
50.根据本发明的一种优选实施方式,步骤2)焙烧的条件包括:于80℃至380℃以下,优选340-380℃下焙烧4~10小时,然后于高于380℃至650℃以下,优选580-650℃下焙烧3~7小时,然后于高于650℃至900℃以下,优选800-880℃焙烧3~6小时;采用前述焙烧方式,使稀土ce富集高效分布在催化剂的表面,有利于在烷基芳烃脱氢生产烷烯基芳烃反应中降低副产物苯的含量。
51.根据本发明的一种优选实施方式,步骤2)干燥的条件包括:室温~80℃,优选50-70℃,干燥的时间可以依据干燥温度进行合理确定,优选干燥时间为3-5h。
52.根据本发明的一种优选实施方式,所述步骤1)中水的用量为步骤1)和步骤2)原料总重量的15-30重量%,优选为步骤1)和步骤2)原料总重量的20-28重量%;采用前述制备方法,有利于稀土ce的分散,使催化剂表面ce的分布更均匀,有利于在烷基芳烃脱氢生产烷
烯基芳烃反应中降低副产物苯的含量。
53.根据本发明的一种优选实施方式,所述步骤1)接触的时间为4-24h。
54.根据本发明的一种优选实施方式,稀土组分r化合物为r盐、r氢氧化物、r氧化物中的一种或多种,进一步优选为可溶性r盐;更优选为硝酸盐。
55.根据本发明的一种优选实施方式,稀土组分铈化合物为铈盐、铈氢氧化物、铈氧化物中的一种或多种,更优选为硝酸铈、草酸铈、碳酸铈中的一种或多种;采用前述铈化合物,有利于稀土ce富集高效分布在催化剂的表面。
56.根据本发明的一种优选实施方式,稀土组分铈化合物为硝酸铈和草酸铈的混合物,以ceo2计,硝酸铈与草酸铈的重量比为0.3-2∶1。
57.根据本发明的一种优选实施方式,形成碱性条件的碱性物质为尿素和/或氨。
58.根据本发明的一种优选实施方式,形成碱性条件的碱性物质的用量为步骤1)和步骤2)原料总重量的1-3重量%。
59.根据本发明的一种实施方式,本发明提供一种所述的制备方法制备得到的稀土氧化物催化剂。
60.本发明中,所述铁源、钾源、可选地其他金属助剂源与粘结剂源的物质种类无特殊要求,本领域常用的种类均可以用于本发明,本发明在此不再赘述。
61.根据本发明,优选所述铁源为氧化铁红和氧化铁黄的混合物,优选以fe2o3计,氧化铁红与氧化铁黄的重量比为2-5∶1。
62.本发明中,捏合例如湿捏、成型等的步骤和条件可以采用本领域常规的步骤和条件,本发明在此不再赘述。
63.本发明中,成型例如可以采用挤条成型。
64.本发明制备的催化剂颗粒可以依据需要成型为实心圆柱形、空心圆柱形、三叶形、菱形、梅花形、蜂窝型等各种形状,其直径和颗粒长度也没有固定的限制。
65.根据本发明的一种优选实施方式,所述催化剂可以成型为直径为3毫米、长5~10毫米的实心圆柱形颗粒,采用前述规格的催化剂有利于在烷基芳烃脱氢生产烷烯基芳烃反应中降低副产物苯的含量。
66.根据本发明的一种优选实施方式,本发明提供本发明所述的催化剂在烷基苯脱氢制备烷烯基中的应用,优选所述烷烯基芳烃为苯乙烯、甲基苯乙烯和二乙烯基苯中的一种或多种。本发明的催化剂特别适合于烷基苯脱氢,特别适合于乙苯脱氢制备苯乙烯,本发明的催化剂能够降低副产物苯的含量。
67.按前述方法制得的催化剂在等温式固定床中进行活性评价,例如对乙苯脱氢制苯乙烯催化剂活性评价而言,过程简述如下:
68.将反应原料分别经计量泵输入预热混合器,预热混合成气态后进入反应器,反应器采用电热丝加热,使之达到预定温度。反应器内径为1
″
的不锈钢管,内可装填100毫升催化剂。由反应器流出的反应物经水冷凝后用气相色谱仪分析其组成。
69.乙苯转化率、苯乙烯选择性、苯选择性按以下公式计算:
70.[0071][0072][0073]
本发明中催化剂的icp-aes测定:采用美国瓦里安公司的varian 725-es仪器,经微波消解后,稀释后进行定性和定量测试。本发明中催化剂的sem-eds测定:采用日本joelo的jsm-35c型扫描电镜,电压为10kv,其中样品表面元素定量分析采用x射线能量谱仪。
[0074]
本发明的催化剂具有副产物苯含量低的特点,使用本发明的催化剂,在反应压力常压、液体空速1.0小时-1
、620℃、水蒸气/乙苯(重量比)2.0条件下,用于乙苯脱氢制苯乙烯的反应,其副产物苯的选择性低至0.35%,取得了较好的技术效果。
[0075]
下面通过实施例对本发明作进一步阐述。
[0076]
实施例1
[0077]
将相当于0.24份y2o3的硝酸钇用占步骤1)和2)总原料总重量20.5%的水进行溶解,加入相当于步骤1)和2)总原料总重量1.2%的尿素混合均匀,然后加入相当于2.02份ceo2的硝酸铈和相当于6.08份ceo2的草酸铈混合均匀,室温下静置6小时得催化剂前体i。
[0078]
将相当于62.27份fe2o3的氧化铁红、相当于12.45份fe2o3的氧化铁黄、相当于13.32份k2o的碳酸钾、相当于1.28份moo3的钼酸铵、相当于1.19份cao的碳酸钙、1.14份mno2、0.02份tio2和6.1份羧甲基纤维素钠在捏合机中搅拌1.3小时,然后加入催化剂前体i,湿捏0.7小时,取出挤条,挤成直径3毫米、长5毫米的颗粒,放入烘箱,50℃烘5小时,然后在340℃下升温焙烧6小时,620℃下焙烧5小时,然后在850℃下焙烧5小时得成品催化剂。催化剂的组成、icp和eds表征结果见表1。
[0079]
将乙苯和水在等温床反应器中,与催化剂进行反应,在反应温度为620℃,反应压力为常压,液体空速为1.0小时-1
,水蒸气/乙苯的重量比为2.0条件下进行评价,测试结果列于表2。
[0080]
实施例2
[0081]
将相当于0.42份y2o3的硝酸钇用占步骤1)和2)总原料总重量24.5%的水进行溶解,加入相当于步骤1)和2)总原料1.6%的尿素混合均匀,然后加入相当于2.31份ceo2的硝酸铈和相当于6.93份ceo2的草酸铈混合均匀,室温下静置24小时得催化剂前体i。
[0082]
将相当于57.91份fe2o3的氧化铁红、相当于14.48份fe2o3的氧化铁黄、相当于11.54份k2o的碳酸钾、相当于2.04份moo3的钼酸铵、相当于2.27份cao的碳酸钙、2.06份mno2、0.05份tio2和5.1份羧甲基纤维素钠在捏合机中搅拌1.5小时,然后加入催化剂前体i,湿捏0.8小时,取出挤条,挤成直径3毫米、长5毫米的颗粒,放入烘箱,55℃烘5小时,然后在370℃下升温焙烧6小时,640℃下焙烧5小时,然后在880℃下焙烧3小时得成品催化剂。催化剂的组成、icp和eds表征结果见表1。
[0083]
将乙苯和水在等温床反应器中,与催化剂进行反应,在反应温度为620℃,反应压力为常压,液体空速为1.0小时-1
,水蒸气/乙苯的重量比为2.0条件下进行评价,测试结果列
于表2。
[0084]
实施例3
[0085]
将相当于0.22份y2o3的硝酸钇用占步骤1)和2)总原料总重量26.5%的水进行溶解,加入相当于步骤1)和2)总原料总重量1.4%的尿素混合均匀,然后加入相当于6.54份ceo2的硝酸铈和相当于3.31份ceo2的草酸铈混合均匀,室温下静置18小时得催化剂前体i。
[0086]
将相当于58.94份fe2o3的氧化铁红、相当于14.74份fe2o3的氧化铁黄、相当于12.21份k2o的碳酸钾、相当于1.91份moo3的钼酸铵、相当于1.05份cao的碳酸钙、1.00份mno2、0.08份tio2和5.8份羧甲基纤维素钠在捏合机中搅拌1.3小时,然后加入催化剂前体i,湿捏0.6小时,取出挤条,挤成直径3毫米、长5毫米的颗粒,放入烘箱,65℃烘5小时,然后在370℃下升温焙烧4小时,650℃下焙烧7小时,然后在800℃下焙烧5小时得成品催化剂。催化剂的组成、icp和eds表征结果见表1。
[0087]
将乙苯和水在等温床反应器中,与催化剂进行反应,在反应温度为620℃,反应压力为常压,液体空速为1.0小时-1
,水蒸气/乙苯的重量比为2.0条件下进行评价,测试结果列于表2。
[0088]
实施例4
[0089]
将相当于0.75份y2o3的硝酸钇用占步骤1)和2)总原料总重量20.5%的水进行溶解,加入相当于步骤1)和2)总原料总重量1.2%的尿素混合均匀,然后加入相当于1.90份ceo2的硝酸铈和相当于5.69份ceo2的草酸铈混合均匀,室温下静置6小时得催化剂前体i。
[0090]
将相当于62.27份fe2o3的氧化铁红、相当于12.45份fe2o3的氧化铁黄、相当于13.32份k2o的碳酸钾、相当于1.28份moo3的钼酸铵、相当于1.19份cao的碳酸钙、1.14份mno2、0.02份tio2和6.1份羧甲基纤维素钠在捏合机中搅拌1.3小时,然后加入催化剂前体i,湿捏0.7小时,取出挤条,挤成直径3毫米、长5毫米的颗粒,放入烘箱,50℃烘5小时,然后在340℃下升温焙烧6小时,620℃下焙烧5小时,然后在850℃下焙烧5小时得成品催化剂。催化剂的组成、icp和eds表征结果见表1。
[0091]
将乙苯和水在等温床反应器中,与催化剂进行反应,在反应温度为620℃,反应压力为常压,液体空速为1.0小时-1
,水蒸气/乙苯的重量比为2.0条件下进行评价,测试结果列于表2。
[0092]
实施例5
[0093]
将相当于0.13份y2o3的硝酸钇用占步骤1)和2)总原料总重量20.5%的水进行溶解,加入相当于步骤1)和2)总原料总重量1.2%的尿素混合均匀,然后加入相当于2.05份ceo2的硝酸铈和相当于6.17份ceo2的草酸铈混合均匀,室温下静置6小时得催化剂前体i。
[0094]
将相当于62.27份fe2o3的氧化铁红、相当于12.45份fe2o3的氧化铁黄、相当于13.32份k2o的碳酸钾、相当于1.28份moo3的钼酸铵、相当于1.19份cao的碳酸钙、1.14份mno2、0.02份tio2和6.1份羧甲基纤维素钠在捏合机中搅拌1.3小时,然后加入催化剂前体i,湿捏0.7小时,取出挤条,挤成直径3毫米、长5毫米的颗粒,放入烘箱,50℃烘5小时,然后在340℃下升温焙烧6小时,620℃下焙烧5小时,然后在850℃下焙烧5小时得成品催化剂。催化剂的组成、icp和eds表征结果见表1。
[0095]
将乙苯和水在等温床反应器中,与催化剂进行反应,在反应温度为620℃,反应压力为常压,液体空速为1.0小时-1
,水蒸气/乙苯的重量比为2.0条件下进行评价,测试结果列
于表2。
[0096]
实施例6
[0097]
将相当于0.06份y2o3的硝酸钇用占步骤1)和2)总原料总重量20.5%的水进行溶解,加入相当于步骤1)和2)总原料总重量1.2%的尿素混合均匀,然后加入相当于0.87份ceo2的硝酸铈和相当于2.63份ceo2的草酸铈混合均匀,室温下静置6小时得催化剂前体i。
[0098]
将相当于65.43份fe2o3的氧化铁红、相当于13.08份fe2o3的氧化铁黄、相当于13.32份k2o的碳酸钾、相当于1.28份moo3的钼酸铵、相当于1.19份cao的碳酸钙、1.14份mno2、0.02份tio2和6.1份羧甲基纤维素钠在捏合机中搅拌1.3小时,然后加入催化剂前体i,湿捏0.7小时,取出挤条,挤成直径3毫米、长5毫米的颗粒,放入烘箱,50℃烘5小时,然后在340℃下升温焙烧6小时,620℃下焙烧5小时,然后在850℃下焙烧5小时得成品催化剂。催化剂的组成、icp和eds表征结果见表1。
[0099]
将乙苯和水在等温床反应器中,与催化剂进行反应,在反应温度为620℃,反应压力为常压,液体空速为1.0小时-1
,水蒸气/乙苯的重量比为2.0条件下进行评价,测试结果列于表2。
[0100]
实施例7
[0101]
按照实施例1的组成和方法制备催化剂、然后评价催化剂,唯一不同的是焙烧为一步焙烧,具体焙烧条件为850℃焙烧16小时。
[0102]
实施例8
[0103]
将相当于0.24份y2o3的硝酸钇用占步骤1)和2)总原料总重量20.5%的水进行溶解,加入相当于步骤1)和2)总原料总重量1.2%的尿素混合均匀,然后加入相当于2.02份ceo2的硝酸铈和相当于6.08份ceo2的草酸铈混合均匀,室温下静置6小时得催化剂前体i。
[0104]
将相当于62.27份fe2o3的氧化铁红、相当于12.45份fe2o3的氧化铁黄、相当于13.32份k2o的碳酸钾、相当于1.28份moo3的钼酸铵、相当于1.19份cao的碳酸钙、1.14份mno2、0.02份tio2和6.1份羧甲基纤维素钠在捏合机中搅拌1.3小时,然后加入催化剂前体i,湿捏0.7小时,取出挤条,挤成直径3毫米、长5毫米的颗粒,放入烘箱,50℃烘5小时,然后在340℃下升温焙烧6小时,620℃下焙烧5小时,然后在850℃下焙烧5小时得成品催化剂。催化剂的组成、icp和eds表征结果见表1。
[0105]
将乙苯和水在等温床反应器中,与催化剂进行反应,在反应温度为620℃,反应压力为常压,液体空速为1.0小时-1
,水蒸气/乙苯的重量比为2.0条件下进行评价,测试结果列于表2。
[0106]
实施例9
[0107]
将0.24份y2o3加入占步骤1)和2)总原料总重量20.5%的水,之后加入相当于步骤1)和2)总原料总重量1.2%的尿素混合均匀,然后加入相当于2.02份ceo2的硝酸铈和相当于6.08份ceo2的草酸铈混合均匀,室温下静置6小时得催化剂前体i。
[0108]
将相当于62.27份fe2o3的氧化铁红、相当于12.45份fe2o3的氧化铁黄、相当于13.32份k2o的碳酸钾、相当于1.28份moo3的钼酸铵、相当于1.19份cao的碳酸钙、1.14份mno2、0.02份tio2和6.1份羧甲基纤维素钠在捏合机中搅拌1.3小时,然后加入催化剂前体i,湿捏0.7小时,取出挤条,挤成直径3毫米、长5毫米的颗粒,放入烘箱,50℃烘5小时,然后在340℃下升温焙烧6小时,620℃下焙烧5小时,然后在850℃下焙烧5小时得成品催化剂。催化
剂的组成、icp和eds表征结果见表1。
[0109]
将乙苯和水在等温床反应器中,与催化剂进行反应,在反应温度为620℃,反应压力为常压,液体空速为1.0小时-1
,水蒸气/乙苯的重量比为2.0条件下进行评价,测试结果列于表2。
[0110]
实施例10
[0111]
将相当于0.24份y2o3的硝酸钇用占步骤1)和2)总原料总重量20.5%的水进行溶解,加入相当于步骤1)和2)总原料总重量1.2%的尿素混合均匀,然后加入相当于2.02份ceo2的硝酸铈和相当于6.08份ceo2的草酸铈混合均匀,室温下静置6小时得催化剂前体i。
[0112]
将相当于62.27份fe2o3的氧化铁红、相当于12.45份fe2o3的氧化铁黄、相当于13.32份k2o的碳酸钾、相当于1.28份moo3的钼酸铵、相当于1.19份cao的碳酸钙和6.1份羧甲基纤维素钠在捏合机中搅拌1.3小时,然后加入催化剂前体i,湿捏0.7小时,取出挤条,挤成直径3毫米、长5毫米的颗粒,放入烘箱,50℃烘5小时,然后在340℃下升温焙烧6小时,620℃下焙烧5小时,然后在850℃下焙烧5小时得成品催化剂。催化剂的组成、icp和eds表征结果见表1。
[0113]
将乙苯和水在等温床反应器中,与催化剂进行反应,在反应温度为620℃,反应压力为常压,液体空速为1.0小时-1
,水蒸气/乙苯的重量比为2.0条件下进行评价,测试结果列于表2。
[0114]
实施例11
[0115]
将相当于0.21份la2o3的硝酸镧用占步骤1)和2)总原料总重量18.2%的水进行溶解,加入相当于步骤1)和2)总原料总重量1.5%的尿素混合均匀,然后加入相当于2.56份ceo2的硝酸铈和相当于7.70份ceo2的草酸铈混合均匀,室温下静置4小时得催化剂前体i。
[0116]
将相当于48.85份fe2o3的氧化铁红、相当于24.43份fe2o3的氧化铁黄、相当于10.20份k2o的碳酸钾、相当于3.60份moo3的钼酸铵、相当于0.49份cao的氢氧化钙、1.76份mno2、0.20份tio2和7.5份羧甲基纤维素钠在捏合机中搅拌1.5小时,然后加入催化剂前体i,湿捏0.6小时,取出挤条,挤成直径3毫米、长5毫米的颗粒,放入烘箱,60℃烘4小时,然后在360℃下升温焙烧8小时,630℃下焙烧4小时,然后在820℃下焙烧6小时得成品催化剂。催化剂的组成、icp和eds表征结果见表1。
[0117]
将乙苯和水在等温床反应器中,与催化剂进行反应,在反应温度为620℃,反应压力为常压,液体空速为1.0小时-1
,水蒸气/乙苯的重量比为2.0条件下进行评价,测试结果列于表2。
[0118]
实施例12
[0119]
将相当于0.49份sc2o3的硝酸钪用占步骤1)和2)总原料总重量25.4%的水进行溶解,加入相当于步骤1)和2)总原料总重量2.1%的尿素混合均匀,然后加入相当于9.73份ceo2的硝酸铈混合均匀,室温下静置5小时得催化剂前体i。
[0120]
将相当于52.16份fe2o3的氧化铁红、相当于17.39份fe2o3的氧化铁黄、相当于15.48份k2o的碳酸钾、相当于0.80份moo3的钼酸铵、相当于3.61份cao的氢氧化钙、0.27份mno2、0.08份tio2和6.8份羧甲基纤维素钠在捏合机中搅拌1.4小时,然后加入催化剂前体i,湿捏0.6小时,取出挤条,挤成直径3毫米、长5毫米的颗粒,放入烘箱,70℃烘3小时,然后在380℃下升温焙烧4小时,580℃下焙烧6小时,然后在800℃下焙烧6小时得成品催化剂。催化
剂的组成、icp和eds表征结果见表1。
[0121]
将乙苯和水在等温床反应器中,与催化剂进行反应,在反应温度为620℃,反应压力为常压,液体空速为1.0小时-1
,水蒸气/乙苯的重量比为2.0条件下进行评价,测试结果列于表2。
[0122]
对比例1
[0123]
将相当于2.08份ceo2的硝酸铈和相当于6.25份ceo2的草酸铈混合均匀用占原料总重量20.5%的水进行溶解,加入相当于原料总量1.2%的尿素混合均匀,室温下静置6小时得催化剂前体i。
[0124]
将相当于62.27份fe2o3的氧化铁红、相当于12.45份fe2o3的氧化铁黄、相当于13.32份k2o的碳酸钾、相当于1.28份moo3的钼酸铵、相当于1.19份cao的碳酸钙、1.14份mno2、0.02份tio2和6.1份羧甲基纤维素钠在捏合机中搅拌1.3小时,然后加入催化剂前体i,湿捏0.7小时,取出挤条,挤成直径3毫米、长5毫米的颗粒,放入烘箱,50℃烘5小时,然后在340℃下升温焙烧6小时,620℃下焙烧5小时,然后在850℃下焙烧5小时得成品催化剂。催化剂的组成、icp和eds表征结果见表1。
[0125]
将乙苯和水在等温床反应器中,与催化剂进行反应,在反应温度为620℃,反应压力为常压,液体空速为1.0小时-1
,水蒸气/乙苯的重量比为2.0条件下进行评价,测试结果列于表2。
[0126]
对比例2
[0127]
将相当于62.27份fe2o3的氧化铁红、相当于12.45份fe2o3的氧化铁黄、相当于13.32份k2o的碳酸钾、相当于0.24份y2o3的硝酸钇、相当于2.02份ceo2的硝酸铈、相当于6.08份ceo2的草酸铈、相当于1.28份moo3的钼酸铵、相当于1.19份cao的碳酸钙、1.14份mno2、0.02份tio2和6.1份羧甲基纤维素钠在捏合机中搅拌1.3小时,然后占原料总重量20.5%的水湿捏0.7小时,取出挤条,挤成直径3毫米、长5毫米的颗粒,放入烘箱,50℃烘5小时,然后在340℃下升温焙烧6小时,620℃下焙烧5小时,然后在850℃下焙烧5小时得成品催化剂。催化剂的组成、icp和eds表征结果见表1。
[0128]
将乙苯和水在等温床反应器中,与催化剂进行反应,在反应温度为620℃,反应压力为常压,液体空速为1.0小时-1
,水蒸气/乙苯的重量比为2.0条件下进行评价,测试结果列于表2。
[0129]
对比例3
[0130]
将相当于0.24份y2o3的硝酸钇用占原料总重量20.5%的水进行溶解,加入相当于原料总重量1.2%的草酸混合均匀,然后加入相当于2.02份ceo2的硝酸铈和相当于6.08份ceo2的草酸铈混合均匀,室温下静置6小时得催化剂前体i。
[0131]
将相当于62.27份fe2o3的氧化铁红、相当于12.45份fe2o3的氧化铁黄、相当于13.32份k2o的碳酸钾、相当于1.28份moo3的钼酸铵、相当于1.19份cao的碳酸钙、1.14份mno2、0.02份tio2和6.1份羧甲基纤维素钠在捏合机中搅拌1.3小时,然后加入催化剂前体i,湿捏0.7小时,取出挤条,挤成直径3毫米、长5毫米的颗粒,放入烘箱,50℃烘5小时,然后在340℃下升温焙烧6小时,620℃下焙烧5小时,然后在850℃下焙烧5小时得成品催化剂。催化剂的组成、icp和eds表征结果见表1。
[0132]
将乙苯和水在等温床反应器中,与催化剂进行反应,在反应温度为620℃,反应压
力为常压,液体空速为1.0小时-1
,水蒸气/乙苯的重量比为2.0条件下进行评价,测试结果列于表2。
[0133]
对比例4
[0134]
将相当于0.08份y2o3的硝酸钇用占原料总重量20.5%的水进行溶解,加入相当于原料总重量1.2%的尿素混合均匀,然后加入相当于2.06份ceo2的硝酸铈和相当于6.19份ceo2的草酸铈混合均匀,室温下静置6小时得催化剂前体i。
[0135]
将相当于62.27份fe2o3的氧化铁红、相当于12.45份fe2o3的氧化铁黄、相当于13.32份k2o的碳酸钾、相当于1.28份moo3的钼酸铵、相当于1.19份cao的碳酸钙、1.14份mno2、0.02份tio2和6.1份羧甲基纤维素钠在捏合机中搅拌1.3小时,然后加入催化剂前体i,湿捏0.7小时,取出挤条,挤成直径3毫米、长5毫米的颗粒,放入烘箱,50℃烘5小时,然后在340℃下升温焙烧6小时,620℃下焙烧5小时,然后在850℃下焙烧5小时得成品催化剂。催化剂的组成、icp和eds表征结果见表1。
[0136]
将乙苯和水在等温床反应器中,与催化剂进行反应,在反应温度为620℃,反应压力为常压,液体空速为1.0小时-1
,水蒸气/乙苯的重量比为2.0条件下进行评价,测试结果列于表2。
[0137]
对比例5
[0138]
将相当于1.55份y2o3的硝酸钇用占原料总重量20.5%的水进行溶解,加入相当于原料总重量1.2%的尿素混合均匀,然后加入相当于1.69份ceo2的硝酸铈和相当于5.09份ceo2的草酸铈混合均匀,室温下静置6小时得催化剂前体i。
[0139]
将相当于62.27份fe2o3的氧化铁红、相当于12.45份fe2o3的氧化铁黄、相当于13.32份k2o的碳酸钾、相当于1.28份moo3的钼酸铵、相当于1.19份cao的碳酸钙、1.14份mno2、0.02份tio2和6.1份羧甲基纤维素钠在捏合机中搅拌1.3小时,然后加入催化剂前体i,湿捏0.7小时,取出挤条,挤成直径3毫米、长5毫米的颗粒,放入烘箱,50℃烘5小时,然后在340℃下升温焙烧6小时,620℃下焙烧5小时,然后在850℃下焙烧5小时得成品催化剂。催化剂的组成、icp和eds表征结果见表1。
[0140]
将乙苯和水在等温床反应器中,与催化剂进行反应,在反应温度为620℃,反应压力为常压,液体空速为1.0小时-1
,水蒸气/乙苯的重量比为2.0条件下进行评价,测试结果列于表2。
[0141]
表1
[0142][0143][0144]
表2
[0145]
性能乙苯转化率%苯乙烯选择性%副产物苯选择性%实施例176.5296.100.35实施例275.9895.630.47实施例376.3795.270.43实施例476.6994.910.68实施例575.5395.470.57实施例671.2595.360.52实施例774.3192.310.85实施例874.3894.360.72实施例975.9395.510.46实施例1075.0195.140.63
实施例1175.1695.360.51实施例1276.0295.130.58对比例172.2193.170.89对比例274.0394.620.78对比例374.8792.471.29对比例474.2193.870.83对比例574.5692.131.31
[0146]
采用稀土组分复配的方法,并且使得稀土ce组分能够富集在催化剂的表面,可以有效降低脱氢产物中副产物苯的选择性,从而降低装置的生产成本。
[0147]
以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1