一种多孔吸附催化材料及其制备方法与流程
1.本发明涉及催化技术领域,尤其涉及一种多孔吸附催化材料及其制备方法。
背景技术:
2.催化反应是在催化剂作用下进行的化学反应,包含扩散、吸附、表面反应、脱附等步骤。根据吸附的工作原理,吸附又分为物理吸附和化学吸附。物理吸附是吸附剂和被吸附物质之间通过分子间范德华力产生的吸附;化学吸附是吸附质分子与固体表面原子(或分子)发生电子的转移、交换或共有,形成吸附化学键的吸附。化学吸附是多相催化反应的重要步骤,与物理吸附相比,化学吸附主要有以下特点:
①
吸附所涉及的力与化学键力相当,比范德华力强得多;
②
吸附热近似等于反应热;
③
吸附是单分子层的;
④
有选择性;
⑤
对温度和压力具有不可逆性。另外,化学吸附还常常需要活化能。在复相催化中,多数属于固体表面催化气相反应,它与固体表面吸附紧密相关。在这类催化反应中,至少有一种反应物是被固体表面化学吸附的,而且这种吸附是催化过程的关键步骤。
3.因此开发一种具有优异吸附功能的催化剂具有重要的应用价值。
技术实现要素:
4.有鉴于此,本发明提供了一种吸附性能好的多孔吸附催化材料的制备方法。
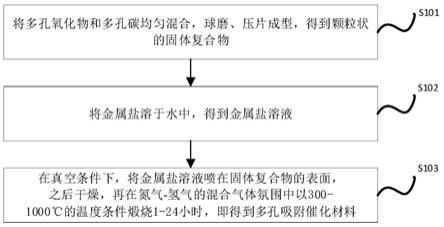
5.本发明提供一种多孔吸附催化材料的制备方法,包括以下步骤:
6.步骤s101,将多孔氧化物和多孔碳均匀混合,球磨、压片成型,得到颗粒状的固体复合物;其中,多孔氧化物与多孔碳的质量比为2:8
‑
8:2;
7.步骤s102,将金属盐溶于水中,得到金属盐溶液;
8.步骤s103,在真空条件下,将金属盐溶液喷在固体复合物的表面,之后干燥,再在氮气
‑
氢气的混合气体氛围中以300
‑
1000℃的温度条件煅烧1
‑
24小时,即得到多孔吸附催化材料;其中,金属盐溶液与固体复合物的质量比为1:5
‑
2:1。
9.进一步地,所述多孔氧化物选用氧化铝、氧化硅或分子筛中的任一种。
10.进一步地,所述多孔碳选用椰壳活性炭。
11.进一步地,所述金属盐选用氯化铜、硫酸铜、硝酸铜、氯化镍、硝酸镍、硫酸镍、氯化钴、硝酸钴或硫酸钴中的任一种或多种。
12.进一步地,所述固体复合物的粒径为1
‑
20毫米。更进一步地,所述固体复合物的粒径为5
‑
12毫米。
13.进一步地,所述多孔吸附催化材料中金属元素的含量为5
‑
80wt%。更进一步地,所述多孔吸附催化材料中金属元素的含量为20
‑
60wt%。
14.进一步地,所述多孔吸附催化材料中多孔碳的含量为1
‑
50wt%。更进一步地,所述多孔吸附催化材料中多孔碳的含量为3
‑
15wt%。
15.进一步地,步骤s103中,煅烧的温度为400
‑
600℃,煅烧时间为2
‑
8小时。
16.进一步地,所述多孔氧化物选用平均孔径为20
‑
30nm的氧化铝;所述多孔碳选用平
均孔径为2
‑
4nm的椰壳活性炭。
17.本发明还提供了利用上述制备方法制得的多孔吸附催化材料。
18.本发明还提供了上述多孔吸附催化材料的应用,该多孔吸附催化材料能够应用于石油化工、煤化工、天然气化工、氢能源等领域。
19.本发明提供的技术方案带来的有益效果是:本发明提供的方法先通过球磨使多孔碳和多孔氧化物充分混合,然后喷涂金属盐溶液,最后形成均匀的无机多孔材料,该无机多孔材料具有多孔氧化物和多孔碳的表面性质和微观孔道结构,与纯多孔氧化物载体相比,表面酸碱性被多孔碳稀释,有利于减少催化副反应的发生;与纯多孔碳载体相比,有利于提高载体的机械强度,适用于更加苛刻的反应条件。
附图说明
20.图1是本发明一种多孔吸附催化材料的制备方法的流程示意图。
具体实施方式
21.为使本发明的目的、技术方案和优点更加清楚,下面将结合附图对本发明实施方式作进一步地描述。
22.请参考图1,本发明的实施例提供了一种多孔吸附催化材料的制备方法,包括以下步骤:
23.步骤s101,将多孔氧化物和多孔碳均匀混合,球磨、压片成型,得到粒径为1
‑
20毫米的颗粒状的固体复合物;其中,多孔氧化物选用氧化铝、氧化硅或分子筛中的任一种;多孔碳选用椰壳活性炭;多孔氧化物与多孔碳的质量比为2:8
‑
8:2;
24.步骤s102,将金属盐溶于水中,得到金属盐溶液;其中,金属盐选用氯化铜、硫酸铜、硝酸铜、氯化镍、硝酸镍、硫酸镍、氯化钴、硝酸钴或硫酸钴中的任一种或多种;
25.步骤s103,在真空条件下,将金属盐溶液喷在固体复合物的表面,之后干燥,再在氮气
‑
氢气的混合气体氛围中以300
‑
1000℃的温度条件煅烧1
‑
24小时,即得到多孔吸附催化材料;金属盐溶液与固体复合物的质量比为1:5
‑
2:1;多孔吸附催化材料中,镍、铜或钴的含量为5
‑
80wt%,多孔碳的含量为1
‑
50wt%。
26.下面结合实施例和对比例对本发明提供的多孔吸附催化材料及其制备方法进行详细说明。
27.实施例1:
28.本发明的实施例1提供了一种多孔吸附催化材料的制备方法,包括以下步骤:
29.步骤s101,称取50千克氧化铝粉体和15千克椰壳活性炭置于高速球磨机中运转10分钟,充分混匀后取出,之后加入少量去离子水和起润滑作用的石墨混匀,利用压片机压片成型,得到粒径为3
‑
8毫米的圆柱形颗粒状的固体复合物,记为su
‑
1;使用的氧化铝粉体的比表面积为280平方米/克,平均孔径为20纳米;椰壳活性炭的平均孔径为2纳米;
30.步骤s102,将硝酸镍加入水中溶解,得到镍含量为15wt%的硝酸镍溶液;
31.步骤s103,称取20千克固体复合物su
‑
1放入密闭的、带有加热、转动功能的不锈钢容器中,对不锈钢容器进行抽真空处理,然后将28千克硝酸镍溶液喷在固体复合物su
‑
1的表面,转动30分钟后,开启加热功能,转动并加热至110℃除去水分,最后在含有50%氮气和
50%氢气的混合气体氛围的高温炉中以500℃煅烧6小时,得到多孔吸附催化材料,标记为cat
‑
1。
32.对比例1:
33.本发明的对比例1提供了一种不含有多孔碳的催化材料的制备方法,包括以下步骤:
34.步骤s101,称取50千克氧化铝粉体置于高速球磨机中运转10分钟,充分混匀后取出,之后加入少量去离子水和石墨混匀,用压片机压片成型,得到粒径为3
‑
8毫米的圆柱形颗粒状的氧化铝颗粒,记为su
‑
2;使用的氧化铝粉体的比表面积为280平方米/克,平均孔径为20纳米;
35.步骤s102,将硝酸镍加入水中溶解,得到镍含量为15wt%的硝酸镍溶液;
36.步骤s103,称取20千克氧化铝颗粒su
‑
2放入密闭的、带有加热、转动功能的不锈钢容器中,对不锈钢容器进行抽真空处理,然后将28千克硝酸镍溶液喷在氧化铝颗粒su
‑
2的表面,转动30分钟后,开启加热功能,转动并加热至110℃除去水分,最后在含有50%氮气和50%氢气的混合气体氛围的高温炉中以500℃煅烧6小时,得到催化材料,记为cat
‑
2。
37.对实施例1和对比例1制得的催化材料进行催化反应评价:
38.一、采用高压釜评价,反应温度为60
‑
150℃,氢气气氛压力为4.0mpa,采用丙酮为探针分子,用量为500克,实施例1和对比例1的催化材料的用量均为10克,反应时间为6小时,反应后取样色谱分析,色谱柱db
‑
wax,检测器fid,该反应的目的是判断催化剂的活性和选择性好坏,目标产物是异丙醇,副产物是甲基异丁基甲酮。
39.分析结果见表1。
40.表1:催化反应评价结果
[0041][0042]
从表1中的结果可以看出,本发明实施例1制得的催化材料cat
‑
1的反应活性和选择性均优于传统的催化材料cat
‑
2,与传统氧化物载体催化剂相比具有显著的技术进步。
[0043]
二、采用高压釜评价,反应温度60
‑
150℃,氢气气氛压力为4.0mpa,采用正丙醛为探针分子,用量为500克,实施例1和对比例1的催化材料的用量均为10克,反应时间为6小时,反应后取样色谱分析,色谱柱db
‑
wax,检测器fid。该反应的目的是判断催化剂的活性和选择性好坏,目标产物是正丙醇,副产物考查丙醚。
[0044]
分析结果见表2。
[0045]
表2:催化反应评价结果
[0046][0047]
从表2中结果可以看出,本发明实施例1制得的催化材料cat
‑
1的反应活性和选择性均优于传统的催化材料cat
‑
2,与传统氧化物载体催化剂相比具有显著的技术进步。
[0048]
三、采用高压釜评价,反应温度60
‑
150℃,氢气气氛压力为4.0mpa,采用正丁醛为探针分子,用量为500克,实施例1和对比例1的催化材料的用量均为10克,反应时间为6小时,反应后取样色谱分析,色谱柱db
‑
wax,检测器fid。该反应的目的是判断催化剂的活性和选择性好坏,目标产物是正丁醇,副产物考查丁醚。
[0049]
分析结果见表3。
[0050]
表3:催化反应评价结果
[0051][0052]
从表3中结果可以看出,本发明实施例1制得的催化材料cat
‑
1的反应活性和选择性均优于传统的催化材料cat
‑
2,与传统氧化物载体催化剂相比具有显著的技术进步。
[0053]
四、采用高压釜评价,反应温度80
‑
150℃,氢气气氛压力为4.0mpa,采用丙酮为探针分子,用量为500克,实施例1和对比例1的催化材料的用量均为10克,反应时间为6小时,反应后取样色谱分析,色谱柱db
‑
wax,检测器fid。该反应的目的是判断催化剂的活性和选择性好坏,目标产物是异丙醇,副产物考查丙醚。
[0054]
分析结果见表4。
[0055]
表4:催化反应评价结果
[0056][0057]
从表4中结果可以看出,本发明实施例1制得的催化材料cat
‑
1的反应活性和选择性均优于传统的催化材料cat
‑
2,与传统氧化物载体催化剂相比具有显著的技术进步。
[0058]
实施例2:
[0059]
本发明的实施例2提供了一种多孔吸附催化材料的制备方法,包括以下步骤:
[0060]
步骤s101,称取40千克氧化硅粉体和30千克椰壳活性炭置于高速球磨机中运转10分钟,充分混匀后取出,之后加入少量去离子水和石墨混匀,利用压片机压片成型,得到粒径为5
‑
10毫米的圆柱形颗粒状的固体复合物;椰壳活性炭的平均孔径为2纳米;
[0061]
步骤s102,将硝酸铜加入水中溶解,得到铜含量为10wt%的硝酸铜溶液;
[0062]
步骤s103,称取15千克固体复合物放入密闭的、带有加热、转动功能的不锈钢容器中,对不锈钢容器进行抽真空处理,然后将12千克硝酸铜溶液喷在固体复合物的表面,转动30分钟后,开启加热功能,转动并加热至110℃除去水分,最后在含有50%氮气和50%氢气的混合气体氛围的高温炉中以600℃煅烧5小时,得到多孔吸附催化材料。
[0063]
实施例3:
[0064]
本发明的实施例3提供了一种多孔吸附催化材料的制备方法,包括以下步骤:
[0065]
步骤s101,称取35千克分子筛和35千克椰壳活性炭置于高速球磨机中运转10分钟,充分混匀后取出,之后加入少量去离子水和石墨混匀,利用压片机压片成型,得到粒径为4
‑
9毫米的圆柱形颗粒状的固体复合物;椰壳活性炭的平均孔径为2纳米;
[0066]
步骤s102,将硝酸钴加入水中溶解,得到钴含量为12wt%的硝酸钴溶液;
[0067]
步骤s103,称取25千克固体复合物放入密闭的、带有加热、转动功能的不锈钢容器中,对不锈钢容器进行抽真空处理,然后将18千克硝酸钴溶液喷在固体复合物的表面,转动30分钟后,开启加热功能,转动并加热至110℃除去水分,最后在含有50%氮气和50%氢气的混合气体氛围的高温炉中以650℃煅烧6小时,得到多孔吸附催化材料。
[0068]
实施例4:
[0069]
本发明的实施例4提供了一种多孔吸附催化材料的制备方法,包括以下步骤:
[0070]
步骤s101,称取60千克氧化铝粉体和20千克椰壳活性炭置于高速球磨机中运转10分钟,充分混匀后取出,之后加入少量去离子水和石墨混匀,利用压片机压片成型,得到粒径为9
‑
12毫米的圆柱形颗粒状的固体复合物;氧化铝粉体的平均孔径为25纳米;椰壳活性炭的平均孔径为3纳米;
[0071]
步骤s102,将氯化镍加入水中溶解,得到镍含量为18wt%的氯化镍溶液;
[0072]
步骤s103,称取40千克固体复合物放入密闭的、带有加热、转动功能的不锈钢容器
中,对不锈钢容器进行抽真空处理,然后将30千克氯化镍溶液喷在固体复合物的表面,转动30分钟后,开启加热功能,转动并加热至110℃除去水分,最后在含有50%氮气和50%氢气的混合气体氛围的高温炉中以700℃煅烧6小时,得到多孔吸附催化材料。
[0073]
实施例5:
[0074]
本发明的实施例5提供了一种多孔吸附催化材料的制备方法,包括以下步骤:
[0075]
步骤s101,称取55千克氧化硅粉体和60千克椰壳活性炭置于高速球磨机中运转10分钟,充分混匀后取出,之后加入少量去离子水和石墨混匀,利用压片机压片成型,得到粒径为9
‑
12毫米的圆柱形颗粒状的固体复合物;椰壳活性炭的平均孔径为3纳米;
[0076]
步骤s102,将氯化钴加入水中溶解,得到钴含量为15wt%的氯化钴溶液;
[0077]
步骤s103,称取30千克固体复合物放入密闭的、带有加热、转动功能的不锈钢容器中,对不锈钢容器进行抽真空处理,然后将25千克氯化钴溶液喷在固体复合物的表面,转动30分钟后,开启加热功能,转动并加热至110℃除去水分,最后在含有50%氮气和50%氢气的混合气体氛围的高温炉中以800℃煅烧6小时,得到多孔吸附催化材料。
[0078]
实施例6:
[0079]
本发明的实施例6提供了一种多孔吸附催化材料的制备方法,包括以下步骤:
[0080]
步骤s101,称取65千克氧化铝粉体和25千克椰壳活性炭置于高速球磨机中运转10分钟,充分混匀后取出,之后加入少量去离子水和石墨混匀,利用压片机压片成型,得到粒径为8
‑
10毫米的圆柱形颗粒状的固体复合物;氧化铝粉体的平均孔径为30纳米;椰壳活性炭的平均孔径为4纳米;
[0081]
步骤s102,将氯化铜加入水中溶解,得到铜含量为16wt%的氯化铜溶液;
[0082]
步骤s103,称取40千克固体复合物放入密闭的、带有加热、转动功能的不锈钢容器中,对不锈钢容器进行抽真空处理,然后将30千克氯化铜溶液喷在固体复合物的表面,转动30分钟后,开启加热功能,转动并加热至110℃除去水分,最后在含有50%氮气和50%氢气的混合气体氛围的高温炉中以850℃煅烧6小时,得到多孔吸附催化材料。
[0083]
在不冲突的情况下,本文中上述实施例及实施例中的特征可以相互结合。
[0084]
以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1