一种高载量的金属单原子催化剂及其制备方法和应用
1.本发明属于功能材料制备技术领域,更具体地,涉及一种高载量的金属单原子催化剂及其制备方法和应用。
背景技术:
2.纳米材料由于其特殊的属性,包括表面与界面效应,量子效应,宏观量子隧道效应等,在过去的二十年间得到了研究者们的广泛关注。在催化领域,纳米材料的催化活性很大程度上来源于其表面存在的大量不饱和配位原子。因此,研究者们通过调控纳米颗粒的尺寸、形貌、晶面等去调控催化剂表面原子的分布和结构,以提高其催化性能。随着纳米催化的发展和表征技术的进步,研究者发现,当纳米颗粒的尺寸降低到团簇,甚至单原子时,其能级结构和电子结构会发生极大的变化,使单原子催化剂往往表现出不同于传统纳米催化剂的催化性能。另外,与纳米粒子相比,单原子催化剂具有最大的原子利用效率,可以完全将活性中心带到反应表面,进一步增强催化活性,并为实现金属资源的合理利用和实现原子经济提供了巨大的潜力。
3.2011年,中科院大连化物所张涛课题组成功制备了单原子pt/feo
x
催化剂,并首次提出了“单原子催化”的概念(nat.chem.2011,3,634
‑
640)。该催化剂在co氧化和co选择性氧化反应中表现出很高的催化活性和稳定性。随后,金属单原子催化剂由于其均匀的金属活性中心,独特的电子结构,理论上100%的金属原子利用效率等特点,在电催化产氢、氧还原、co2转化等领域得到了广泛关注和研究。除了优异的催化性能,单原子材料结构的简单性和均质性有助于活性位点的精确识别和表征,为从分子层次认识催化反应的机理提供了理想的模型和研究平台,并有助于在原子尺度上为目标反应实现合理的催化剂设计。单原子催化剂有望成为具有工业催化应用潜力的新型催化剂。
4.尽管金属单原子催化剂具有诸多优势,其面临的一个主要挑战是单原子活性位点的低浓度,这很大程度上限制了其催化活性。由于高表面能,孤立的单个金属原子很容易迁移和聚集成粒子。另外,金属单原子催化剂的制备通常需要高温(例如超过700℃),而在高温条件下金属原子更容易迁移和聚集。因此,在实际的合成和反应条件下制备单原子催化剂并保持金属原子的分散性,尤其在高金属负载量的情况下保持单原子活性位点非常具有挑战性。在金属单原子催化剂中,金属氮碳单原子催化剂备受关注,尤其在电催化领域。这是由于氮不仅可以有效地固定和稳定碳上的单个金属原子,而且可以调节金属和碳原子的电子结构。此外,碳载体具有很高的导电性,有利于反应过程中的电子转移。
技术实现要素:
5.为了解决上述现有技术存在的不足和缺点,本发明目的在于提供一种高载量的金属单原子催化剂的制备方法。该方法是通过有机席夫碱反应制备富氮多孔聚合物;利用多孔聚合物中丰富的活性氮原子锚定金属原子;最后通过高温碳化得到高载量金属单原子催化剂(简写为sam
‑
nc);金属以金属
‑
氮
‑
碳(简写为m
‑
n
‑
c)的键合方式在富氮多孔碳载体上
呈原子级分散。
6.本发明的另一目的在于提供上述方法制得的高载量的金属单原子催化剂。该催化剂兼具高比表面积。
7.本发明的目的通过下述技术方案来实现:
8.一种高载量的金属单原子催化剂的制备方法,包括如下具体步骤:
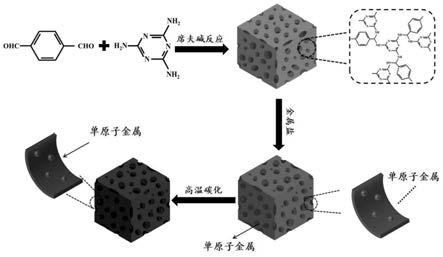
9.s1.将三聚氰胺、对苯二甲醛和二甲基亚砜,惰性氛围下在175~180℃加热回流并搅拌,反应结束后将反应体系冷却至室温,产物经过滤和洗涤,然后在60~80℃下真空干燥去除溶剂,得到富氮多孔聚合物,简写为snw
‑
1;
10.s2.将snw
‑
1和金属乙酸盐加入到乙醇中超声,然后将分散体在50~90℃真空条件干燥得到粉末,简写为snw
‑
1@m粉末,其中m为金属原子;
11.s3.将snw
‑
1@m粉末在惰性气氛下600~900℃碳化,经去离子水洗涤,真空干燥后,制得高载量的金属单原子催化剂。
12.优选地,步骤s1中所述三聚氰胺的物质的量、对苯二甲醛的物质的量和二甲基亚砜的体积比为(2~3)mmol:(3~4)mmol:(10~20)ml。
13.更为优选地,所述三聚氰胺的物质的量、对苯二甲醛的物质的量和二甲基亚砜的体积比为2.485mmol:3.728mmol:15.5ml。
14.优选地,步骤s2中所述snw
‑
1和金属乙酸盐的质量比为1:(0.05~0.2);所述snw
‑
1和金属乙酸盐的总质量与乙醇的体积比为0.55g:(30~120)ml。
15.更为优选地,所述金属乙酸盐为乙酸钴、乙酸镍、乙酸铁、乙酸钼、乙酸钯、乙酸铜或乙酸锰中的一种以上。
16.优选地,步骤s3中所述惰性气氛为氮气;所述金属单原子催化剂中金属的载量为5~8wt%。
17.优选地,步骤s1中所述搅拌的时间为48~96h,步骤s2中所述超声的时间为0.5~4h;所述干燥的时间为9~48h;步骤s3中所述碳化的时间为0.5~3h。
18.一种高载量的金属单原子催化剂,所述金属单原子催化剂是由所述的方法制备得到。
19.优选地,所述高载量的金属单原子催化剂的比表面积为300~850m
2 g
‑1;孔体积为0.2~0.5cm
3 g
‑1。
20.所述的高载量的金属单原子催化剂在吸附、储能或催化领域中的应用。
21.本发明利用三聚氰胺和对苯二甲醛发生席夫碱反应制备富氮多孔聚合物,将富氮多孔聚合物与金属乙酸盐通过超声充分混合并反应,然后利用富氮多孔聚合物丰富的活性n原子锚定金属(m),最后在惰性气氛中高温碳化得到高载量的金属单原子催化剂。所制备的金属单原子催化剂具有以下几个显著特点:一是金属单原子催化剂中金属的载量为5~8wt%,高载量的金属原子有利于提高催化剂的性能;第二个显著特点是金属以m
‑
n
‑
c的方式在碳载体上呈原子级分散,这保证了金属原子的稳定性,以及金属原子的利用效率;第三个显著特点是材料的表面积和孔体积分别300~850m
2 g
‑1和(0.2~0.5cm
3 g
‑1),这有利于催化活性位点的裸露,以及催化反应中的传质。
22.与现有技术相比,本发明具有以下有益效果:
23.1.本发明通过三聚氰胺和对苯二甲醛发生席夫碱反应制备富氮多孔聚合物。该多
孔聚合物具有丰富活性氮原子和高比表面积(300~850m
2 g
‑1)和孔体积(0.2~0.5cm
3 g
‑1)。富氮多孔聚合物中大量的氮原子有利于锚定大量的金属原子,使金属单原子催化剂中金属的载量为5~8wt%。
24.2.本发明利用制备的富氮多孔聚合物中丰富的氮原子锚定金属。通过snw
‑
1和金属盐反应,可使金属原子被锚定在活性氮原子上,得到snw
‑
1@m材料。
25.3.本发明高温碳化制备高载量金属单原子催化剂。将snw
‑
1@m粉末在惰性气氛下600
‑
900℃碳化可得到最终产物为金属单原子催化剂sam
‑
nc,该材料具有高比表面积。
附图说明
26.图1为本发明的技术路线示意图。
27.图2为实施例1得到的saco
‑
nc和单纯富氮碳材料nc的xrd图谱。
28.图3为实施例1得到的saco
‑
nc的氮气吸脱附等温线和孔径分布图片。
29.图4为实施例1制得的saco
‑
nc的sem和元素能量分布面描分析(eds)的照片。
30.图5为实施例1制得的saco
‑
nc的tem和元素能量分布面描分析(eds)的照片。
31.图6为添加实施例1制得的saco
‑
nc后的li2s6溶液和空白样纯li2s6溶液分别在静置3h的可视化照片及紫外
‑
可见吸收光谱。
32.图7为实施例1制得的saco
‑
nc的x射线光电子能谱(xps)图片。
33.图8为实施例1制得的saco
‑
nc的拉曼光谱照片。
34.图9为实施例2制得的sapd
‑
nc的sem和元素能量分布面描分析(eds)照片。
35.图10为实施例2得到的sapd
‑
nc的xrd图谱。
36.图11为实施例2得到的sapd
‑
nc的氮气吸脱附等温线和孔径分布图片。
具体实施方式
37.下面结合具体实施例进一步说明本发明的内容,但不应理解为对本发明的限制。若未特别指明,实施例中所用的技术手段为本领域技术人员所熟知的常规手段。除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备。
38.实施例1
39.图1为本发明的技术路线示意图。首先,利用三聚氰胺和对苯二甲醛发生席夫碱反应制备富氮多孔聚合物(snw
‑
1);然后,将snw
‑
1与钴乙酸盐通用超声充分混合并反应,以利用snw
‑
1中丰富的氮原子锚定金属;最后,通过在惰性气氛中高温碳化得到高钴载量的金属单原子催化剂(saco
‑
nc)。具体步骤如下所示:
40.1.制备:
41.(1)在三口烧瓶中装入三聚氰胺(313mg,2.485mmol),对苯二甲醛(500mg,3.728mmol)和二甲基亚砜(15.5ml),在惰性氛围下加热回流并搅拌。加热温度为180℃,持续72h。反应结束后,将反应体系冷却至室温,过滤得到固体粉末,然后用过量的丙酮,四氢呋喃和二氯甲烷洗涤固体产物,最后在室温下真空去除溶剂,得到呈黄白色粉末状的snw
‑
1材料(收率为61%)。
42.(2)将0.5g snw
‑
1材料和0.05g钴乙酸盐加入到50ml乙醇中,超声分散2小时,然后将分散体在70℃真空条件干燥24小时得到粉末snw
‑
1@co。
43.(3)将snw
‑
1@co粉末在惰性气氛下800℃碳化1h,得到黑色粉末。经去离子水洗涤,真空干燥后得到高载量的金属单原子催化剂,简写为saco
‑
nc。
44.2.性能表征:
45.图2为实施例1得到的saco
‑
nc和单纯富氮碳材料nc的xrd图谱。两种材料的xrd图谱均在约25
°
处出现一个宽峰,及在约44
°
处出现一个弱峰,均是碳材料的特征峰。对于saco
‑
nc材料,没有任何钴的结晶峰出现,表明没有结晶钴的形成,也从侧面反映出金属钴的原子级分散。图3为实施例1得到的saco
‑
nc的氮气吸脱附等温线和孔径分布图片。其中,(a)为氮气吸脱附等温线,(b)为孔径分布。可以看出,saco
‑
nc材料具有多孔(微孔
‑
介孔共存)结构,平均孔径约为2nm。图4为实施例1制得的saco
‑
nc的sem和元素能量分布面描分析(eds)照片。其中,(a)为saco
‑
nc形貌,(b)为c元素分布,(c)为n元素分布,(d)为co元素分布,从图中(b)
‑
(d)可以看出,碳(c),氮(n)和钴(co)原子在多孔碳载体中高度均匀地分散。图5为实施例1制得的saco
‑
nc的tem和元素能量分布面描分析(eds)照片。其中,(a)为saco
‑
nc形貌,(b)为c元素分布,(c)为n元素分布,(d)为co元素分布,从图中(b)
‑
(d)可以看出,碳(c),氮(n)和钴(co)原子在多孔碳载体中高度均匀地分散。图6为添加实施例1制得的saco
‑
nc后的li2s6溶液和空白样纯li2s6溶液分别在静置3h的可视化照片及紫外
‑
可见吸收光谱。从图6中可以看出,空白的多硫化物li2s6观察到深黄色,而含有saco
‑
nc的溶液在3小时后变得无色;420nm处的uv
‑
vis吸收峰反映了li2s6的浓度,在混合多硫化物溶液和saco
‑
nc后,多硫化物li2s6的峰变得非常弱,说明saco
‑
nc对多硫化锂有强吸附作用,可将其用于锂硫电池领域抑制多硫化物穿梭效应。图7为saco
‑
nc的x射线光电子能谱(xps)图片。其中,(a)为n1s的xps核心光谱,(b)为co 2p的xps核心光谱,(c)为saco
‑
nc的xps调查光谱。可以看出,saco
‑
nc中的氮是吡啶
‑
氮、吡咯
‑
氮、石墨
‑
氮和钴
‑
氮的形式。吡啶、吡咯和石墨氮原子的发现意味着氮被掺杂到碳中,而钴
‑
氮形式的存在,表明钴与氮的键合作用。图8为实施例1得到的saco
‑
nc和nc拉曼测试的图片,图中表现出拉曼光谱表现出一阶g频段(≈1610cm
‑1)和d频段(≈1310cm
‑1),是氮碳材料的典型拉曼峰。
46.上述saco
‑
nc中co的载量达到5.51wt%以上,高载量的金属原子有利于提高催化剂的性能;金属co以co
‑
n
‑
c的方式在碳载体上呈原子级分散,这保证了金属原子的稳定性,以及金属原子的利用效率;材料的比表面积和孔体积分别达728.1m
2 g
‑1和0.369cm
3 g
‑1以上,这有利于催化活性位点的裸露以及催化反应中的传质。
47.实施例2
48.利用三聚氰胺和对苯二甲醛发生席夫碱反应制备富氮多孔聚合物(snw
‑
1);然后,将snw
‑
1与钯乙酸盐通用超声充分混合并反应,以利用snw
‑
1中丰富的氮原子锚定金属;最后,通过在惰性气氛中高温碳化得到高钯载量的金属单原子催化剂(sapd
‑
nc)。具体步骤如下所示:
49.1.制备:
50.(1)在三口烧瓶中装入三聚氰胺(313mg,2.485mmol),对苯二甲醛(500mg,3.728mmol)和二甲基亚砜(15.5ml),在惰性氛围下加热回流并搅拌。加热温度为180℃,持续72h。反应结束后,将反应体系冷却至室温,过滤得到固体粉末,然后用过量的丙酮,四氢呋喃和二氯甲烷洗涤固体产物,最后在室温下真空去除溶剂,得到呈黄白色粉末状的snw
‑
1材料(收率为61%)。
51.(2)将0.5g snw
‑
1材料和0.1g钯乙酸盐加入到50ml乙醇中,超声分散2小时,然后将分散体在70℃真空条件干燥24小时得到粉末snw
‑
1@pd。
52.(3)将snw
‑
1@co粉末在惰性气氛下800℃碳化1h,得到黑色粉末。经去离子水洗涤,真空干燥后得到高载量的金属单原子催化剂,简写为sapd
‑
nc。
53.2.性能表征:图9为实施例2制得的sapd
‑
nc的sem和元素能量分布面描分析(eds)照片。其中,(a)为sapd
‑
nc形貌,(b)为c元素分布,(c)为n元素分布,(d)为pd元素分布。从图中(b
‑
(d)中可以看出,碳(c),氮(n)和钯(pd)原子在多孔碳载体中高度均匀地分散,其中金属钯载量为5.26wt%以上。图10为实施例2得到的sapd
‑
nc的xrd图谱。从图10可知,在约25
°
处出现一个宽峰,及在约44
°
处出现一个弱峰,均是碳材料的特征峰。对于sapd
‑
nc材料,没有任何钯的结晶峰出现,表明没有结晶钯的形成,也从侧面反映出金属钯的原子级分散。图11为实施例2得到的sapd
‑
nc的氮气吸脱附等温线和孔径分布图片。其中,(a)为氮气吸脱附等温线,(b)为孔径分布。可以看出,sapd
‑
nc材料具有多孔(微孔
‑
介孔共存)结构。
54.上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合和简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1