一种适于低温催化氧化富氧燃烧烟气汞的催化剂及其制备方法
1.本发明涉及一种适于低温催化氧化富氧燃烧烟气汞的催化剂及其制备方法,属于大气污染净化技术领域。
背景技术:
2.燃煤电厂co2排放是全球变暖的一个重要因素。为了在2050年前将地球表面的平均温度升高控制在2
‑
2.4℃之间,co2排放量必须减少50
‑
85%。co2捕获和储存(ccs)是减少煤炭燃烧过程中co2排放的有效方法,而富氧燃烧(o2/co2燃烧)是目前技术上可行、经济上最具竞争力的ccs方法之一。与空气燃烧(o2/n2燃烧)相比,因用纯氧气/循环烟气代替o2/n2燃烧煤粉,富氧燃烧烟气成分发生了较大改变:co2浓度达95%以上;h2o、so2、so3和hg浓度均成倍增加。富氧燃烧烟气中,除了颗粒物、so2和no
x
等常规污染物需脱除外,由于烟气中的汞会与铝制co2压缩装置发生汞齐反应,生成铝汞齐,使压缩设备脆化,造成重大安全事故,因此汞在进入co2压缩装置之前必须从烟气中脱除。
3.在煤燃烧过程中,汞主要以单质汞(hg0)、颗粒态汞(hg
p
)和二价汞(hg
2+
)三种形式存在于燃煤烟气中。其中,二价汞(hg
2+
)因易溶于水而容易被湿法脱硫装置(wfgd)脱除,颗粒态汞(hg
p
)很容易随颗粒物被静电除尘器或布袋除尘器等除尘设备除去,而单质汞(hg0)由于其强挥发性以及水不溶性,现有的烟气净化装置很难将其脱除。已有研究结果表明利用氧化剂或催化剂将hg0转化为hg
2+
,再借助湿法脱硫装置(wfgd)脱除可实现hg0的有效控制。o2/n2燃烧中,30~80%hg0可被电厂配置的选择性催化还原(scr)脱硝系统催化氧化,因此scr催化剂协同脱除hg/no已经成为o2/n2燃烧下的研究热点。o2/co2燃烧中,因no
x
脱除需求,仍要配置scr系统,若采用空气燃烧电厂中的高温scr系统布置方式,烟气中通过scr过程形成的n2又被循环烟气带入高温炉膛,其在高温燃烧过程会再次氧化成no
x
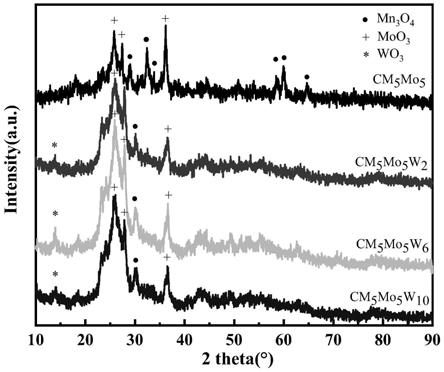
,部分削弱scr系统的作用,将scr系统配置于富氧燃烧烟气循环取点(温度<150℃)之后的布置方式则能有效避免上述问题,且该布置方式烟气温度降低,体积缩小,催化剂用量减少,经济性增强。所以研究富氧燃烧低温scr催化剂协同脱除hg/no具有重要现实意义,而适于富氧燃烧烟气成分的低温催化剂开发是关键。
4.已有研究表明mno
x
是一种性能良好的低温催化剂活性成分,在100
‑
250℃温度范围内,mn
4+
能从吸附于催化剂表面的hg0获得电子,自身被还原成mn
3+
或mn
2+
,hg0则会被氧化成hg
2+
。烟气中o2则能吸附于催化剂表面活性位点形成晶格氧,再将mn
3+
或mn
2+
氧化成mn
4+
,实现汞的低温高效催化氧化,但是不良的mno
x
结构会抑制催化剂的活性,通过在多孔载体上负载mno
x
可以有效改善mno
x
的氧化性能。
5.在众多催化剂载体材料中,因碳基材料表面负载金属氧化物制备出的催化剂因同时具备碳基材料的高吸附性和金属氧化物的氧化性而备受关注。碳纳米管(cnt)是一种新型一维纳米碳基材料,其内部类石墨层隙结构能提供大量吸附位点,bet比表面积可高达2000m2/g,且cnt石墨化程度高,具备较强的电子转移能力,能通过快速电子转移机制强化
催化反应过程。研究结果显示将mno
x
负载于cnt表面合成的mn/cnt在200℃达到了90%以上的汞氧化效率。但是mn/cnt的抗硫特性较差,当烟气中存在500ppmso2时,其汞氧化效率降低至30%。在富氧燃烧中,由于烟气再循环,烟气中so2浓度会成倍增加。
技术实现要素:
6.本发明的技术目的旨在结合富氧燃烧烟气的特点,为富氧燃烧烟气中单质汞的催化氧化提供一种性能优异的低温催化剂,填补富氧燃烧中汞氧化催化剂的空白。该催化剂的活性成分为二氧化锰,助剂成分为三氧化钼和三氧化钨,载体成分为多壁碳纳米管,通过调节催化剂中活性成分及助剂的比例,使得催化剂具备较高的低温汞催化氧化汞效率,并能有效增强催化剂的抗硫特性。
7.本发明的技术构思是采用助剂mo可提高活性组分的分散性,mo
6+
对so2的亲和性高于mn
4+
,且mo
6+
能强化o2对低价态金属活性成分的氧化,有效阻断so2与活性组分之间的直接接触,从而提高mn/cnt催化剂的抗硫特性。另外,在催化剂中添加w有效利用富氧燃烧烟气中co2含量较高的特点,基于wo3制备的纳米管具备大量表面w
‑
o空位结构,能形成稳定的电子传输通道,常温下可将co2转化为c
‑
o等活性基团,有效增强催化剂反应活性。基于此,本发明拟在mn/cnt中添加助剂mo和w,以此合成一种适于低温催化氧化富氧燃烧烟气汞的催化剂。
8.本发明为实现技术目的采用的技术方案是:一种适于低温催化氧化富氧燃烧烟气汞的催化剂,其特征在于:所述催化剂包括质量百分比为5%~10%的二氧化锰,质量百分比为5%~8%的三氧化钼,质量百分比为2%~10%的三氧化钨,所述催化剂中的其余成分为多壁碳纳米管。
9.进一步地,所述催化剂被研磨成80~325目。
10.一种适于低温催化氧化富氧燃烧烟气汞的催化剂的制备方法,包括以下步骤:
11.(1)将80~88重量份的多壁碳纳米管与多壁碳纳米管质量20倍的去离子水混合,在室温下搅拌均匀,得到混合物ai;
12.(2)按步骤(1)中去离子水的1/5体积取浓度为10wt%的hno3添加至混合物ai中,在室温下搅拌均匀,得到混合物aii;
13.(3)按步骤(1)中去离子水的1/10体积取浓度为10wt%柠檬酸添加至混合物aii中,在室温下搅拌均匀,得到混合物aiii;
14.(4)称取10.29~20.58重量份的mn(no3)2溶于去离子水得溶液bi;称取5~8重量份的三氧化钼,溶解于与步骤(2)中的hno3等体积的氨水中,得到溶液bii;称取2.187~10.936重量份的钨酸铵,溶解于体积为步骤(1)中去离子水的1/5的去离子水中,得到溶液biii;
15.(5)将步骤(4)得到的溶液bi、bii和biii先后导入混合物aiii中,然后加入浓硝酸调节ph值至1.0,并在50℃条件下震荡24h,然后升温至80℃并在80℃条件下干燥48h,得到固体a;
16.(6)将步骤(5)得到的固体a置于马弗炉中,升温至400℃,在惰性气氛下煅烧5h,研磨,得所述的催化剂。
17.优先地,所述步骤(6)中,固体a按照10℃/min的升温速率升温。
18.目前对于富氧燃烧烟气中汞的催化氧化仍然集中于已在传统空气燃烧中应用的高温钒基催化剂,由于富氧燃烧中的烟气再循环,高温钒基催化剂已经不再适用,因此需要能够配置于静电除尘器之后的低温催化剂,但是有关低温催化剂在富氧燃烧烟气中的汞氧化效果及机理研究的报道较少。
19.本发明的申请人经过研究发现,由于cnt的纳米管道结构和高比表面积,本发明中以cnt为载体制备的锰基催化剂的比表面积均高于170m2/g,远高于传统催化剂的50~70m2/g,在mn/cnt中添加moo3和wo3后,使催化剂表面存在大量mno2,moo3和wo3,说明该种方法合成的催化剂表面的活性成分多以高价态形态存在,具备较强的氧化能力。另外,moo3的添加阻止了so2在催化剂表面吸附,从而阻止了so2进一步向so3的转变,增强了催化剂的抗硫特性。而wo3的添加,则显著增强了催化剂在高浓度co2存在的烟气中的催化活性。
20.本发明的申请人通过大量实验,研究了不同温度和不同活性成分添加比例下该催化剂对富氧燃烧烟气单质汞的催化氧化效果。实验结果表明,当催化剂中二氧化锰含量为5%~10%,三氧化钼含量为5%~8%,三氧化钨的含量为2%~10%时,催化剂在so2存在的富氧燃烧烟气条件下的最佳汞氧化效率可以达到94%,因此,在合理的活性成分配比下,能够制备出一种适于在低温条件下催化氧化富氧燃烧烟气单质汞的催化剂,有效弥补富氧燃烧烟气中低温汞氧化催化剂开发的空白。
附图说明
21.图1为不同w添加量催化剂样品的xps分析:(a)mn 2p;
22.图2为不同w添加量催化剂样品的xps分析:(b)mo 3d;
23.图3为不同w添加量催化剂样品的xps分析:(c)w 4f;
24.图4为本发明制得的催化剂的xrd分析图。
具体实施方式
25.下面结合具体实施例对本发明作进一步具体详细描述,但本发明的实施方式不限于此,对于未特别注明的工艺参数,可参照常规技术进行。本发明实施例中所用多壁碳纳米管均购自于深圳市穗恒科技有限公司(牌号为cas308068
‑
56
‑
6)。
26.实施例1催化剂(cm5mo5w2)的制备
27.按以下步骤:
28.(1)将88重量份的多壁碳纳米管与多壁碳纳米管质量20倍的去离子水混合,在30℃下搅拌1h,得到混合物ai;
29.(2)按步骤(1)中去离子水的1/5体积取浓度为10wt%的hno3添加至混合物ai中,在30℃下搅拌0.5h,得到混合物aii;
30.本步骤中hno3的主要作用在于将疏水性的多壁碳纳米管链状结构打开,便于浸渍活性成分,使用浓硝酸不行,使用其他浓度的稀hno3不会影响效果
31.(3)按步骤(1)中去离子水的1/10体积取浓度为10wt%柠檬酸添加至混合物aii中,在30℃下搅拌0.5h,得到混合物aiii;
32.本步骤中柠檬酸的作用在于增强后续添加的活性物质在溶液中的分散性,其他浓度的柠檬酸不会影响效果。
33.(4)称取浓度为50wt%的mn(no3)2溶液bi 20.58重量份,称取5重量份的三氧化钼溶解于与步骤(2)中的hno3溶液等体积的氨水中,得到溶液bii;称取2.187重量份的钨酸铵溶解于体积为步骤(1)中去离子水的1/5的去离子水中,得到溶液biii;
34.本步骤中三氧化钼能溶于氨水和强碱,为了不引入其他难处理的杂质成分,所以选择了氨水能溶解三氧化钼,从图1~图3的xps分析结果来看,hno3、柠檬酸、mn(no3)2、钼酸铵、钨酸铵、碳纳米管的添加后改变了活性成分mn和mo在催化剂表面存在的形态和比例,如更高的mn
4+
和mo
6+
含量,这一变化可能使得催化剂利于富氧烟气中汞的氧化。
35.(5)将步骤(4)得到的溶液bi、bii和biii先后导入混合物aiii中,然后加入浓硝酸调节ph值至1.0,并在50℃条件下震荡24h,然后升温至80℃并在80℃条件下干燥48h,得到固体a;
36.只有酸性较强,即ph值较低时才能合成性能较好的催化剂,此处为了煅烧过程中能将杂质成分脱除,因此添加hno3调节ph。
37.(6)将步骤(5)得到的固体a置于马弗炉中,以10℃/min的升温速率升温至400℃,在惰性气氛下煅烧5h,研磨,得本催化剂。
38.该催化剂中二氧化锰的含量为5重量份、三氧化钼的含量为5重量份、三氧化钨的含量为2重量份,其余成分为多壁碳纳米管。
39.实施例2催化剂(cm5mo5w6)的制备
40.按以下步骤:
41.(1)将84重量份的多壁碳纳米管与多壁碳纳米管质量20倍的去离子水混合,在30℃下搅拌1h,得到混合物ai;
42.步骤(2)~(3)与实施例1相同;
43.(4)称取浓度为50wt%的mn(no3)2溶液bi 20.58重量份,称取5重量份的三氧化钼溶解于与步骤(2)中的hno3溶液等体积的氨水中,得到溶液bii;称取6.562重量份的钨酸铵溶解于体积为步骤(1)中去离子水的1/5的去离子水中,得到溶液biii;
44.(5)将步骤(4)得到的溶液bi、bii和biii先后导入混合物aiii中,然后加入浓硝酸调节ph值至1.0,并在50℃条件下震荡24h,然后升温至80℃并在80℃条件下干燥48h,得到固体a;
45.(6)将步骤(5)得到的固体a置于马弗炉中,以10℃/min的升温速率升温至400℃,在惰性气氛下煅烧5h,研磨,得本催化剂。
46.实施例3催化剂(cm5mo5w
10
)的制备
47.按以下步骤:
48.(1)将80重量份的多壁碳纳米管与多壁碳纳米管质量20倍的去离子水混合,在30℃下搅拌1h,得到混合物ai;
49.步骤(2)~(3)与实施例1相同;
50.(4)称取浓度为50wt%的mn(no3)2溶液bi 20.58重量份,称取5重量份的三氧化钼溶解于与步骤(2)中的hno3溶液等体积的氨水中,得到溶液bii;称取10.936重量份的钨酸铵溶解于体积为步骤(1)中去离子水的1/5的去离子水中,得到溶液biii;
51.(5)将步骤(4)得到的溶液bi、bii和biii先后导入混合物aiii中,然后加入浓硝酸调节ph值至1.0,并在50℃条件下震荡24h,然后升温至80℃并在80℃条件下干燥48h,得到
固体a;
52.(6)将步骤(5)得到的固体a置于马弗炉中,以10℃/min的升温速率升温至400℃,在惰性气氛下煅烧5h,研磨,得本催化剂。
53.对比实施例1催化剂(cm5)的制备
54.按以下步骤:
55.(1)将95重量份的多壁碳纳米管与多壁碳纳米管质量20倍的去离子水混合,在30℃下搅拌1h,得到混合物ai;
56.步骤(2)~(3)与实施例1相同;
57.(4)称取浓度为50wt%的mn(no3)2溶液bi 20.58重量份;
58.(5)将步骤(4)得到的溶液bi导入混合物aiii中,然后加入浓硝酸将调节ph值至1.0,并在50℃条件下震荡24h,然后升温至80℃并在80℃条件下干燥48h,得到固体a;
59.(6)将步骤(5)得到的固体a置于马弗炉中,以10℃/min的升温速率升温至400℃,在惰性气氛下煅烧5h,研磨,得本催化剂。
60.对比实施例2催化剂(cm5mo5)的制备
61.按以下步骤:
62.(1)将90重量份的多壁碳纳米管与多壁碳纳米管质量20倍的去离子水混合,在30℃下搅拌1h,得到混合物ai;
63.步骤(2)~(3)与实施例1相同;
64.(4)称取浓度为50wt%的mn(no3)2溶液bi 20.58重量份;称取5重量份的三氧化钼,溶解于步骤(2)中的hno3等体积的氨水中,得到溶液bii;
65.(5)将步骤(4)得到的溶液bi和bii先后导入混合物aiii中,然后加入浓硝酸将调节ph值至1.0,并在50℃条件下震荡24h,然后升温至80℃并在80℃条件下干燥48h,得到固体a;
66.(6)将步骤(5)得到的固体a置于马弗炉中,以10℃/min的升温速率升温至400℃,在惰性气氛下煅烧5h,研磨,得本催化剂。
67.催化剂活性测试实验
68.在固定床反应器上分别测试本发明的三个实施例及两个对比实施例制备的六个催化剂的活性,测试项目为汞氧化率。测试条件为:模拟烟气流量为0.9nm3/h;模拟烟气包括以下体积浓度的成分:0.04%no,0.04%nh3,6%o2,8%h2o,0.08%so2,0.002%hcl,和55μg/nm
3 hg0,其余成分为co2;反应温度为50℃,100℃和150℃;单次测试的催化剂用量为0.05g。经过催化剂处理后的烟气采用俄罗斯lumex公司生产的ra915m测汞仪连续在线测试烟气中hg。质量浓度,其测量范围为0~200μg/m3。精度为0.1μg/m3。催化剂氧化hg0的能力通过,hg0氧化率表征,式中:η为hg0氧化率;c0为反应器出口处hg0质量浓度,μg/m3;c1为反应器入口处hg0质量浓度,μg/m3。检测原理为hg0对253.7nm的紫外光有选择性吸收性。测试结果如下表所示:
[0069][0070]
当mno2、moo3的重量百分比低于5%时,脱汞效果极差。提高mno2、moo3的重量百分比可增强脱汞效果,但也增加成本。
[0071]
从上表可以看出:在含有5%mno2和5%moo3的催化剂中添加6%~10%wo3后,能够显著提高催化剂在100℃时富氧燃烧烟气中的单质汞氧化性能,最佳汞氧化效率达94%。
[0072]“改性碳纳米管对富氧燃烧烟气中单质汞的催化氧化特性”介绍了添加mo可以使催化剂抗硫,但是co2浓度过高使活性锰成分碳酸盐化,因此co2体积分数过高会强烈抑制该催化剂对汞的氧化;该篇文献中催化反应的实验温度为150℃,本发明中在添加w后使得催化剂的最佳活性温度进一步降低至100℃,wo3纳米管结构能够促使co2向c
‑
o
‑
c结构转化,在100℃时co2应优先在活性wo3内部转化,从而避免了锰成分的碳酸盐化。
[0073]
由于cnt的纳米管道结构和高比表面积,本发明中以cnt为载体制备的锰基催化剂的比表面积均高于170m2/g,远高于传统催化剂的50~70m2/g,见表1的结果。
[0074]
表1催化剂的bet比表面积、孔体积和孔径分布
[0075][0076]
如附图1~图4所示,在mn/cnt中添加moo3和wo3后,能够明显看到催化剂表面存在大量mno2,moo3和wo3,说明该种方法合成的催化剂表面的活性成分多以高价态形态存在,具备较强的氧化能力。另外,moo3的添加阻止了so2在催化剂表面吸附,从而阻止了so2进一步向so3的转变,增强了催化剂的抗硫特性。而wo3的添加,则显著增强了催化剂在高浓度co2存在的烟气中的催化活性。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1