一种乙烯选择性四聚用催化剂及其制备方法和应用
1.本发明涉及乙烯齐聚反应技术领域,具体涉及一种乙烯选择性四聚用催化剂及其制备方法和应用。
背景技术:
2.线性α-烯烃(lao)作为重要的化工原料,可用于制备润滑油、表面活性剂等,其中1-己烯、1-辛烯是合成线性低密度聚乙烯(lldpe)与高密度聚乙烯(hdpe)中不可或缺的共聚单体(lldpe中共聚单体含量一般为8-10%,hdpe中共聚单体含量为1-2%)。乙烯齐聚作为生产线性α-烯烃的一种重要方法,相较于蜡裂解、煤品提取、萃取分离等传统方法而言,在产品质量上具有很大提升,现已广泛应用于工业化生产当中。
3.传统的乙烯齐聚催化主要使用金属钛系、锆系、铁系等,这些催化体系主要遵循cossee-arlman机理,即乙烯分子插入到催化剂金属中心线性链增长,得到的线性α-烯烃通常呈正态分布,工业应用时须根据需求加以分离纯化。而乙烯高选择齐聚主要遵循金属环化机理,这样生产出来的α-烯烃呈schulz-flory分布,峰值处的产物具有较高的含量,该方法为生产特定碳数的α-烯烃提供了重要的途径。近年来,1-己烯、1-辛烯需求量的增长使得乙烯选择性齐聚又成为工业和学术研究的热点。
4.目前,乙烯高选择性齐聚的报道主要有二聚、三聚、四聚制1-丁烯、1-己烯、1-辛烯。这些催化体系中,催化剂的结构调控对产物分布起到了关键作用,而催化剂结构的调控取决于配体的骨架和取代基的变化。近年来,该领域的研究集中在乙烯选择性齐聚催化机理和配体设计上,并取得了一些重要成果。2002年,british petroleum公司报道了cr/pnp催化体系用于选择性制备1-己烯(chem.commun.2002,858)。2003年,phillips petroleum公司利用开发出的phillips三聚铬催化剂实现了乙烯三聚工业化(us5523507),我国中石化(燕山)和中石油(大庆)也相继采用类似的催化体系实现了工业化生产1-己烯。通过乙烯四聚选择性地制备1-辛烯现在还无法实现工业化生产。而1-辛烯可以用于制备主要应用于生产高品质聚乙烯(pe)、聚烯烃弹性体(poe)、润滑油基础油(pao)、增塑剂、表面活性剂等领域。相对于1-己烯,1-辛烯有更高的经济价值。2008年,sasol公司又合成了一系列稳定性更高的碳桥联双膦配体用于催化乙烯选择性四聚(j.mol.catal.a:chem.2008,283,114)。2010年,韩国sk能源公司开发出一系列双甲基取代、带手性骨架的dppe类型配体用于催化乙烯选择性四聚(organometallics 2010,29,5805),但是活性较低,最高只能达到238kg/(g cr/h)。2013年,zhang等人设计并合成了一系列乙烯基桥联的含双苯基膦基(-pph2)的双膦配体,ph2p(r)c=c(h)pph2,催化乙烯选择性三聚、四聚反应,显现出较好的乙烯三聚和四聚催化活性(acs catal.,2013,3,2311)。但由于有较多1-己烯生成,1-辛烯含量只有50%左右。而且聚合物含量大于1%,易发生聚合物挂壁,缠搅拌桨等现象,影响工业装置的连续运行。聚合物的生成是由于催化剂的稳定性一般,催化剂发生降解的产物使乙烯发生高聚而得到的。
技术实现要素:
5.本发明的目的就是为了克服上述现有技术存在的缺陷而提供一种乙烯选择性四聚用催化剂及其制备方法和应用。
6.本发明的目的可以通过以下技术方案来实现:
7.本发明为进一步提高1-辛烯的选择性和该催化体系的活性,本发明创造性的在乙烯基桥联的双膦配体(ph2p(r)c=c(h)pph2)的磷原子上引入一个或多个烷基替代苯基。通过引入空间位阻较小的烷基,本发明成功将该催化体系的1-辛烯选择性提升至81%。此外,由于烷基的供电子能力比苯基更强,配体中引入烷基使磷原子的电子云密度增大,可显著增加铬金属活性中心的稳定性,延长催化剂寿命,有效提高了该催化体系的活性。催化剂寿命的提高也减少了催化剂的降解,进而使得反应中聚合物的含量降至0.05-0.1%,具体方案如下:
8.一种乙烯选择性四聚用催化剂,该催化剂包括配体、过渡金属化合物以及活化剂,其中,配体的化学结构式如下式(i)所示:
[0009][0010]
式中,基团r1至r2各自独立地为卤素、烃基、被取代的杂烃基、氢、芳基、杂原子芳烃或杂烃基;
[0011]
基团r3至r6各自独立地为脂肪烃基、环烃基、杂烃基、被取代的烃基、被取代的杂烃基、被取代的环烃基、芳基、杂原子芳基、被取代的芳基或被取代的杂原子芳基,且r3至r6中至少一个为脂肪烃基、环烃基、杂烃基、被取代的烃基、被取代的杂烃基或被取代的环烃基;优选脂肪烃基。
[0012]
进一步地,所述的基团r1、r2分别独立地为氢、氟、氯、溴、碘、甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基、仲戊基、异戊基、环戊基、正己基、仲己基、异己基、环己基、正庚基、2-甲基环戊基、2,6-二甲基环己基、环庚基、金刚烷基、甲氧基、乙氧基、异丙氧基、叔丁基氧基、苄基、对甲基苄基、邻甲基苄基、间甲基苄基、对叔丁基苄基、间叔丁基苄基、邻叔丁基苄基、对异丙基苄基、间异丙基苄基、邻异丙基苄基、苯基、对氟苯基、邻氟苯基、间氟苯基、对乙基苯基、邻乙基苯基、间乙基苯基、2,4-二甲基苯基、2,4-二异丙基苯基、2,4-二叔丁基苯基、2,6-二甲基苯基、2,6-二异丙基苯基、3,5-二甲基苯基、3,5-二叔丁基苯基、2,4,6-三甲基苯基、萘基、蒽基、联苯基、2-噻吩基、3-噻吩基。
[0013]
基团r3至r6分别独立地为甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基、仲戊基、异戊基、环戊基、正己基、仲己基、异己基、环己基、正庚基、环庚基、正辛基、正癸基、2-甲基环戊基、2,6-二甲基环己基、金刚烷基、甲氧基、乙氧基、异丙氧基、叔丁基氧基、苄基、对甲基苄基、邻甲基苄基、间甲基苄基、对叔丁基苄基、间叔丁基苄基、邻叔丁基苄基、对异丙基苄基、间异丙基苄基、邻异丙基苄基、苯基、对氟苯基、邻氟苯基、间氟苯基、对乙基苯基、邻乙基苯基、间乙基苯基、2,4-二甲基苯基、2,4-二异丙基苯基、2,4-二叔丁基苯基、2,6-二甲基苯基、2,6-二异丙基苯基、3,5-二甲基苯基、3,5-二叔丁基苯基、2,4,
6-三甲基苯基、萘基、蒽基、联苯基,且r3至r6中至少一个选自甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基、仲戊基、异戊基、环戊基、正己基、仲己基、异己基、环己基、正庚基、环庚基、正辛基、正癸基、2-甲基环戊基、2,6-二甲基环己基、金刚烷基、甲氧基、乙氧基、异丙氧基、叔丁基氧基。
[0014]
优选的,基团r1、r2分别独立地选自氢、氟、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基、环戊基、正己基、环己基、正庚基、2-甲基环戊基、2,6-二甲基环己基、环庚基、金刚烷基、甲氧基、乙氧基、异丙氧基、叔丁基氧基、苄基、对甲基苄基、邻甲基苄基、间甲基苄基、对异丙基苄基、间异丙基苄基、邻异丙基苄基、苯基、对氟苯基、邻氟苯基、间氟苯基、对乙基苯基、邻乙基苯基、间乙基苯基、2,6-二甲基苯基、2,6-二异丙基苯基、萘基、蒽基、联苯基、2-噻吩基、3-噻吩基。
[0015]
优选的,基团r3至r6分别独立地为乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基、环戊基、正己基、环己基、正庚基、环庚基、正辛基、正癸基、2-甲基环戊基、2,6-二甲基环己基、金刚烷基、异丙氧基、叔丁基氧基、苄基、对甲基苄基、邻甲基苄基、间甲基苄基、对叔丁基苄基、间叔丁基苄基、苯基、对氟苯基、邻氟苯基、间氟苯基、对乙基苯基、邻乙基苯基、2,6-二异丙基苯基、3,5-二甲基苯基、3,5-二叔丁基苯基、2,4,6-三甲基苯基、萘基、蒽基、联苯基,且r3至r6中至少一个选自乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基、环戊基、正己基、环己基、正庚基、环庚基、正辛基、正癸基、2-甲基环戊基、2,6-二甲基环己基、金刚烷基、异丙氧基、叔丁基氧基。
[0016]
更优选的,r1、r2分别独立地选自氢、氟、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基、环戊基、正己基、环己基、正庚基、2-甲基环戊基、环庚基、异丙氧基、叔丁基氧基、苄基、苯基、对氟苯基、邻氟苯基、间氟苯基、对乙基苯基、邻乙基苯基、间乙基苯基、2,6-二甲基苯基、2,6-二异丙基苯基、萘基、蒽基、2-噻吩基、3-噻吩基。
[0017]
更优选的,基团r3至r6分别独立地选自乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基、环戊基、正己基、环己基、正庚基、环庚基、异丙氧基、叔丁基氧基、苄基、苯基、对氟苯基、邻氟苯基、间氟苯基、对乙基苯基、邻乙基苯基、间乙基苯基、2,6-二甲基苯基、2,6-二异丙基苯基、萘基、蒽基,且r3至r6中至少一个选自乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基、环戊基、正己基、环己基、正庚基、环庚基、异丙氧基、叔丁基氧基。
[0018]
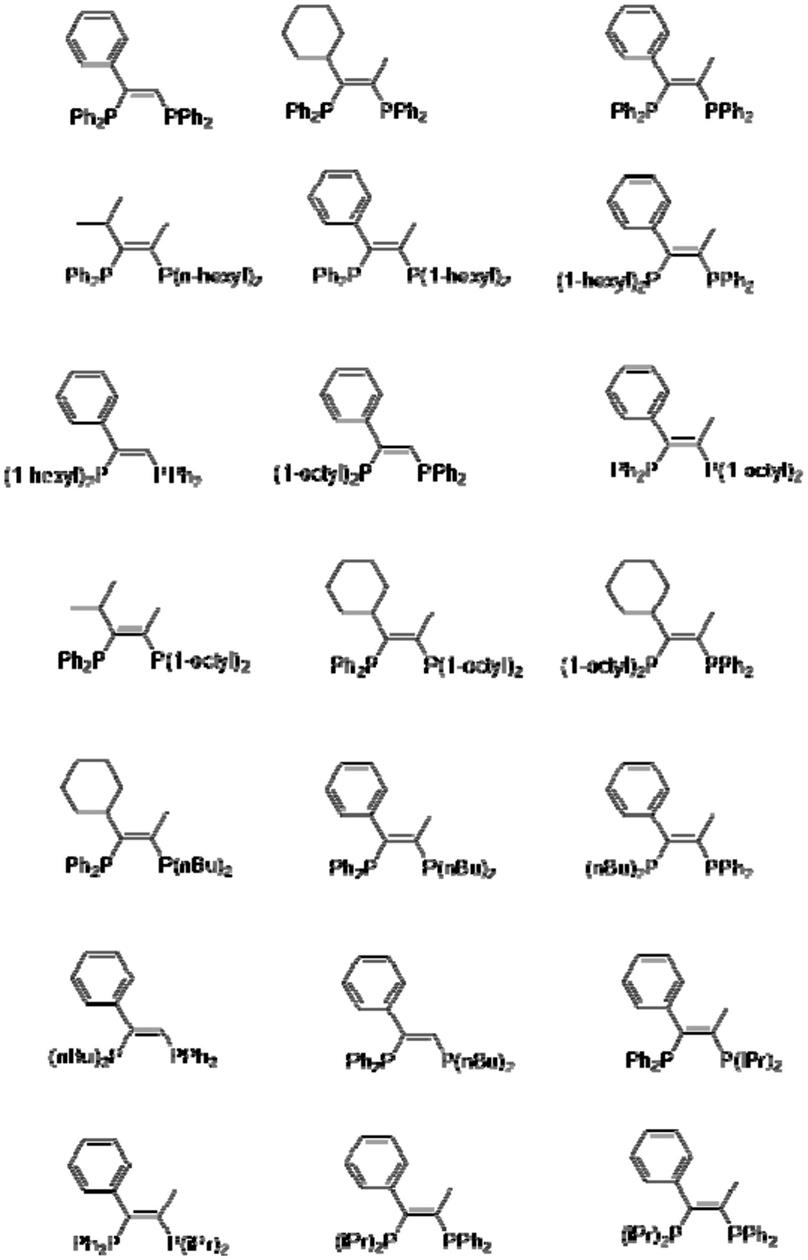
在一些实施方式中,配体化合物为以下物质中的一种,但应理解的是本发明的范围并不限于这些例子:
[0019]
[0020]
[0021][0022]
本发明的催化剂体系中的过渡金属可以是本领域常用的过渡金属化合物,过渡金属化合物中的金属原子为金属活性中心,在催化过程中起重要作用。
[0023]
进一步地,所述的过渡金属化合物中的过渡金属选自铁、钴、镍、铜、钛、钒、铬、锰、钼、钨、镍或钯中的一种。优选的,过渡金属化合物中的过渡金属选自铬、钴、钛、铁、镍或钯中的一种。更优选的,过渡金属化合物中的过渡金属选自铬,具体的,对应的过渡金属化合物为能使低聚进行的任何铬化合物均可以,可选择的铬化合物包括通式crrn所示的化合物,式中rn为有机阴离子或中性分子,rn中一般含有1~15个碳原子,n为0~6的整数,cr的价态在0~6价。具体的rn基团为含羧基、β-二酮基以及烃基的有机物或者其他基团。从易于溶解和易于操作的角度考虑,更适宜的铬化合物包括三氯化铬-三(四氢呋喃)络合物、(苯)三羰基铬、辛酸铬(iii)、六羰基铬、乙酰丙酮铬(iii)、环烷酸铬(iii)、2-乙基己酸铬(iii)、乙酸铬(iii)、2,2,6,6-四甲基庚二酮铬(iii)以及氯化铬(iii)的一种。优选的,铬化合物选自三氯化铬-三(四氢呋喃)络合物、乙酰丙酮铬(iii)、2-乙基己酸铬(iii)。
[0024]
本发明的催化剂体系中的活化剂在催化剂体系中起到活化作用。本发明中可用的活化剂可以是当与配体和过渡金属化合物混合时形成活性催化剂的任意化合物。活化剂可以单独使用或者组合使用。
[0025]
进一步地,所述的活化剂包括烷基铝化合物、铝氧烷化合物、有机硼化合物、无机酸或无机盐中的一种或几种的混合。
[0026]
具体而言,活化剂可以为烷基铝化合物,烷基铝化合物可以为各种三烷基铝,如三甲基铝、三乙基铝、三异丁基铝、三正丁基铝、三正已基铝或三正辛基铝;烷基铝化合物也可以为烷基铝卤化物、烷基铝氢化物或烷基铝倍半氯化物,如一氯二乙基铝(alet2cl)和三氯三乙基二铝(al2et3cl3)。
[0027]
具体而言,活化剂可以为铝氧烷化合物,其通常可以通过将水与烷基铝化合物(例如,三甲基铝)混合而制备得到。所制备的铝氧烷低聚化合物可以是直链化合物、环状化合物、笼状化合物或它们的混合物。合适的铝氧烷化合物可以选自甲基铝氧烷(mao)、乙基铝氧烷、异丁基铝氧烷、改性的铝氧烷和去除挥发性组分的甲基铝氧烷dmao等。
[0028]
具体而言,合适的硼化合物可以包括环硼氧烷、三乙基硼烷、三苯基硼烷、三(五氟苯基)硼烷等。可将有机硼化合物以与有机铝化合物混合的形式使用。
[0029]
优选的,活化剂可以选自甲基铝氧烷(mao)、乙基铝氧烷、异丁基铝氧烷及改性的甲基铝氧烷(mmao)。
[0030]
进一步地,所述的铝氧烷化合物具体包括改性甲基铝氧烷mmao-3a。
[0031]
进一步地,所述的配体与过渡金属化合物中的过渡金属元素的摩尔比为(0.01-100):1,优选(0.1-10):1,更优选(0.5-2):1;
[0032]
所述的活化剂与过渡金属化合物中过渡金属元素的摩尔比为(1-10000):1,优选(1-2000):1,更优选(600-1000):1。
[0033]
一种如上所述乙烯选择性四聚用催化剂的制备方法,该方法为:将配体、过渡金属化合物和活化剂预先混合或直接加入到反应体系中进行原位合成,即得到乙烯选择性四聚用催化剂。
[0034]
下面继续对本发明催化剂体系制备方法做进一步说明:
[0035]
在一些实施方式中,可在存在溶剂或不存在溶剂的条件下,将具有化学式(i)所示的配体、过渡金属化合物与活化剂同时或以任意顺序混合,从而提供活性催化剂。可在-20~250℃下进行催化剂组分的混合,并且在催化剂组分的混合过程中,烯烃的存在通常表现
出保护效果,从而提供了改善的催化性能。进一步而言,可以在约20-100℃的温度范围内进行催化剂组分的混合。
[0036]
在一些实施方式中,可以由过渡金属化合物和化学式(i)所示的配体原位制备可分离的金属-配体络合物。然后将金属-配体络合物加入到反应介质中。可供选择地,可以分别将铬化合物和配体加入到反应器中,由此原位制备铬-配体络合物。原位制备络合物是指在发生催化反应的介质中制备络合物,最后,再添加活化剂。
[0037]
一种如上所述乙烯选择性四聚用催化剂的应用,该催化剂用于乙烯选择性四聚反应生成1-辛烯。
[0038]
进一步地,所述的反应在惰性溶剂中进行,反应的温度为0-200℃,优选10-120℃,更优选20-100℃,进一步优选35-60℃,反应压力为0.1-50mpa,优选1.0-10mpa,优选4-10mpa,过渡金属化合物中的过渡金属在惰性溶剂中的浓度为0.01-10000μmol/l,优选1-500μmol/l。
[0039]
进一步地,所述的惰性溶剂包括烷烃、芳烃、烯烃或离子液体中的一种或几种的混合。典型的溶剂包括但不限于苯、甲苯、二甲苯、异丙苯、氯苯、二氯苯、氟苯、正庚烷、正己烷、甲基环己烷、环己烷、1-己烯、1-辛烯等,优选甲苯、甲基环己烷。
[0040]
与现有技术相比,本发明具有以下优点:
[0041]
(1)本发明创造性的在乙烯基桥联的双膦配体的磷原子上引入一个或多个烷基替代苯基,使得1-辛烯的选择性得到了突破性的提升;
[0042]
(2)本发明在活化剂方面,采用特殊改性后的改性甲基铝氧烷,使催化剂体系在不需要加水的情况下同样达到很高的反应活性;
[0043]
(3)本发明通过优化工艺参数,又可以将1-辛烯的选择性得到进一步提升。
具体实施方式
[0044]
下面对本发明的实施例作详细说明,本实施例在以本发明技术方案为前提下进行实施,给出了详细的实施方式和具体的操作过程,但本发明的保护范围不限于下述的实施例。
[0045]
实施例1
[0046]
(1)配体l1的制备
[0047]
在一个充满氩气的50ml schlenk烧瓶中,加入乙炔基苯(1.0g,10.0mmol)、四氢呋喃(10ml),搅拌并冷却至0℃,将正丁基锂(4.0ml,2.5m己烷溶液,10.0mmol)逐滴加入溶液中,在该温度下搅拌30分钟。然后逐滴加入cy(et)pcl(1.8g,10.0mmol),加完后将混合物升至室温,并搅拌1小时。反应完全后,真空除去挥发物后,混合物用石油醚(30ml)萃取。然后过滤所得混合物以除去不溶性盐,真空干燥滤液,硅胶柱分离纯化,得到白色固体产物(2.4g,98.0%)。
[0048]
在一个充满氩气的schlenk管中,加入上述白色固体产物(0.9g,3.5mmol)、碘化亚铜(34.0mg,0.2mmol)、碳酸铯(114.0mg,0.4mmol)和干燥且脱气的dmf(15ml),搅拌,加入二苯基膦(0.7g,3.9mmol)。将所得混合物在90℃条件下搅拌6小时。冷却至室温后,加入水(20ml),产物用乙酸乙酯(15ml
×
3)萃取。合并的有机层用硫酸钠干燥并减压浓缩,硅胶柱分离纯化,得到白色固体产物l1(0.8g,54.9%)。
[0049]1h nmr(400mhz,cdcl3)δ=7.45-7.40(m,6h),7.37
–
7.29(m,5h),7.18
–
7.15(m,4h),6.66
–
6.60(m,1h),1.50-1.46(m,2h),1.46
–
1.30(m,11h),1.00-0.96(m,3h);
31
p nmr(162mhz,cdcl3)δ=-5.70(d,j=145.6hz),-17.72(d,j=144.6hz).
[0050][0051]
(2)络合物1的制备
[0052]
在一个干燥的并充满氩气的schlenk反应管中,加入配体l1(215.1mg,0.5mmol)和crcl3(thf)3(187.3mg,0.5mmol),向其中加入重蒸二氯甲烷(10ml)室温下搅拌2小时。反应结束后过滤,滤液抽干,得到固体用正己烷洗涤(5ml
×
3),抽干得到蓝色粉末300.0mg(0.46mmol,91%)。
[0053]
实施例2
[0054]
(1)配体l2的制备
[0055]
在一个充满氩气的50ml schlenk烧瓶中,加入乙炔基苯(1.0g,10.0mmol)、重蒸四氢呋喃(10ml),搅拌并冷却至0℃,将正丁基锂(4.0ml,2.5m己烷溶液,10.0mmol)缓慢加入溶液中,在该温度下搅拌30分钟。然后逐滴加入et2pcl(1.2g,10.0mmol),并将混合物升温至环境温度并搅拌1小时。真空除去挥发物后,混合物用石油醚(30ml)萃取。然后过滤所得混合物以除去不溶性盐,真空干燥滤液,硅胶柱分离纯化,得到白色固体产物(1.86g,98.0%)。
[0056]
在一个充满氩气的schlenk管中,加入上述白色固体产物(0.7g,3.50mmol)、碘化亚铜(34mg,0.18mmol)、碳酸铯(114mg,0.35mmol)和干燥且脱气的dmf(15ml),搅拌,加入二苯基膦(0.73g,3.90mmol)。将所得混合物在90℃搅拌6小时。冷却至室温后,加入水(20ml),产物用乙酸乙酯(15ml
×
3)萃取。合并的有机层用硫酸钠干燥并减压浓缩除去溶剂,硅胶柱分离纯化,得到白色固体产物l2(0.73g,55.3%)。
[0057]1h nmr(400mhz,cdcl3)δ=7.44-7.37(m,6h),7.35-7.28(m,5h),7.17-7.13(m,4h),6.56(d,j=34.5hz,1h),1.58-1.50(m,4h),1.00-0.93(m,6h);
31
p nmr(162mhz,cdcl3)δ=-5.51(d,j=163.6hz),-20.49(d,j=164.0hz);
[0058][0059]
(2)络合物2的制备
[0060]
在一个干燥的并充满氩气的schlenk反应管中,加入配体l2(188.1mg,0.5mmol)和crcl3(thf)3(187.3mg,0.5mmol),向其中加入重蒸二氯甲烷(10ml),室温下搅拌2h。反应结束后过滤,滤液抽干,得到固体用正己烷洗涤(5ml
×
3),抽干得到蓝色粉末242.0mg(0.40mmol,80%)。
[0061]
实施例3
[0062]
(1)配体l3的制备
[0063]
在一个充满氩气的50ml schlenk烧瓶中,加入环己基乙炔(1.1g,10.0mmol)和10ml重蒸四氢呋喃,搅拌并冷却至0℃,将正丁基锂(4.00ml,2.5m己烷溶液,10.0mmol)缓慢加入溶液中,在该温度下搅拌30分钟。然后逐滴加入et2pcl(1.2g,10.0mmol),并将混合物升温至环境温度并搅拌1小时。真空除去挥发物后,混合物用石油醚(30ml)萃取。然后过滤所得混合物以除去不溶性盐,真空干燥滤液,硅胶柱分离纯化,得到白色固体产物(1.92g,98.0%)。
[0064]
在一个充满氩气的schlenk管中,加入上述白色固体产物(686.5mg,3.5mmol)、碘化亚铜(34.0mg,0.2mmol)、碳酸铯(114.0mg,0.4mmol)和干燥且脱气的dmf(15ml),搅拌,最后加入二苯基膦(0.7g,3.9mmol)。将所得混合物在90℃搅拌6小时。冷却至室温后,加入水(20ml),产物用乙酸乙酯(15ml
×
3)萃取。合并的有机层用硫酸镁干燥并减压浓缩除去溶剂,得到的残余物硅胶柱分离纯化,得到白色固体产物l3(0.43g,32.0%)。
[0065]1h nmr(400mhz,cdcl3)δ=7.45-7.40(m,6h),7.20-7.15(m,4h),6.00(d,j=29.4hz,1h),1.58-1.50(m,4h),1.50-1.36(m,11h),0.93-0.90(m,6h);
31
p nmr(162mhz,cdcl3)δ=-4.62(d,j=110.7hz),-20.44(d,j=108.6hz);
[0066][0067]
(2)络合物3的制备
[0068]
在一个干燥的并充满氩气的schlenk反应管中,加入配体l3(191.1mg,0.5mmol)和crcl3(thf)3(187.3mg,0.5mmol),向其中加入重蒸二氯甲烷(10ml),室温下搅拌2h。反应结束后过滤,滤液抽干,得到固体用正己烷洗涤(5ml
×
3),抽干得到蓝色粉末268.9mg(0.44mmol,87%)。
[0069]
实施例4
[0070]
(1)配体l4的制备
[0071]
在一个充满氩气的50ml schlenk烧瓶中,加入环己基乙炔(1.1g,10.0mmol)和10ml重蒸四氢呋喃,搅拌并冷却至0℃,将正丁基锂(4.00ml,2.5m己烷溶液,10.0mmol)缓慢加入溶液中,在该温度下搅拌30分钟。然后逐滴加入et2pcl(1.2g,10.0mmol),并将混合物升温至环境温度并搅拌1小时。真空除去挥发物后,混合物用石油醚(30ml)萃取。然后过滤所得混合物以除去不溶性盐,真空干燥滤液,硅胶柱分离纯化,得到白色固体产物(1.92g,98.0%)。
[0072]
在一个充满氩气的schlenk管中,加入上述白色固体产物(686.5mg,3.5mmol)、碘化亚铜(34.0mg,0.2mmol)、碳酸铯(114.0mg,0.4mmol)和干燥且脱气的dmf(15ml),搅拌,最后加入乙基苯基膦(0.54g,3.90mmol)。将所得混合物在90℃搅拌6小时。冷却至室温后,加入水(20ml),产物用乙酸乙酯(15ml
×
3)萃取。合并的有机层用硫酸钠干燥并减压浓缩除去溶剂,硅胶柱分离纯化,得到白色固体产物l4(0.48g,40.6%)。
[0073]1h nmr(400mhz,cdcl3)δ=7.48-7.45(m,3h),7.17
–
7.10(m,2h),5.90(d,j=35.0hz,1h),1.58-1.54(m,4h),1.53-1.50(m,2h),1.50-1.36(m,11h),0.93-0.90(m,6h),0.87-0.83(m,3h);
31
p nmr(162mhz,cdcl3)δ=-6.59(d,j=103.1hz),-26.7(d,j=
110.1hz).
[0074][0075]
(2)络合物4的制备
[0076]
在一个干燥的并充满氩气的schlenk反应管中,加入配体l4(167.1mg,0.5mmol)和crcl3(thf)3(187.3mg,0.5mmol),向其中加入重蒸二氯甲烷(10ml),室温下搅拌2h。反应结束后过滤,滤液抽干,得到固体用正己烷洗涤(5ml
×
3),抽干得到蓝色粉末239.3mg(0.43mmol,85%)。
[0077]
实施例5
[0078]
(1)配体l5的制备
[0079]
参考j.am.chem.soc.,2007,129,4099.中描述的步骤,在一个充满氩气的50ml schlenk烧瓶中,加入环己基乙炔(1.1g,10.0mmol)和10ml重蒸四氢呋喃,搅拌并冷却至0℃,将正丁基锂(4.00ml,2.5m己烷溶液,10.0mmol)缓慢加入溶液中,在该温度下搅拌30分钟。然后逐滴加入ph2pcl(2.21g,10.0mmol),并将混合物升温至环境温度并搅拌1小时。真空除去挥发物后,混合物用石油醚(30ml)萃取。然后过滤所得混合物以除去不溶性盐,真空干燥滤液,得到白色固体产物(2.72g,98.0%)。
[0080]
在一个充满氩气的schlenk管中,加入上述白色固体产物(0.9g,3.5mmol)、碘化亚铜(34.0mg,0.2mmol)、碳酸铯(114.0mg,0.4mmol)和干燥且脱气的dmf(15ml),搅拌,最后加入二苯基膦(0.7g,3.9mmol)。将所得混合物在90℃搅拌6小时。冷却至室温后,加入水(20ml),产物用乙酸乙酯(15ml
×
3)萃取。合并的有机层用硫酸钠干燥并减压浓缩除去溶剂硅胶柱分离纯化,得到白色固体产物l5(1.1g,66.2%)。
[0081]1h nmr(400mhz,cdcl3)δ=7.23-7.37(m,20h),7.03(d,j=36.0hz,1h),2.08-2.02(m,1h),1.49-1.52(m,4h),1.35-1.38(m,2h),1.23-1.31(m,2h),1.13-1.19(m,2h);
31
p nmr(162mhz,cdcl3)δ=-4.08(d,j=166.4hz),-25.83(d,j=166.4hz).
[0082][0083]
(2)络合物5的制备
[0084]
在一个干燥的并充满氩气的schlenk反应管中,加入配体l5(226.1mg,0.5mmol)和crcl3(thf)3(187.3mg,0.5mmol),向其中加入重蒸二氯甲烷(10ml),室温下搅拌2h。反应结束后过滤,滤液抽干,得到固体用正己烷洗涤(5ml
×
3),抽干得到蓝色粉末303.1mg(0.44mmol,89%)。
[0085]
实施例6
[0086]
(1)配体l6的制备
[0087]
在一个充满氩气的50ml schlenk烧瓶中,加入乙炔基苯(1.0g,10.0mmol)和10ml重蒸四氢呋喃,搅拌并冷却至0℃,将正丁基锂(4.00ml,2.5m己烷溶液,10.0mmol)缓慢加入
溶液中,在该温度下搅拌30分钟。然后逐滴加入ph2pcl(2.20g,10.0mmol),并将混合物升温至环境温度并搅拌1小时。真空除去挥发物后,混合物用石油醚(30ml)萃取。然后过滤所得混合物以除去不溶性盐,真空干燥滤液,得到白色固体产物(2.80g,98.0%)。
[0088]
在一个充满氩气的schlenk管中,加入上述白色固体产物(1.0g,3.5mmol)、碘化亚铜(34.0mg,0.2mmol)、碳酸铯(114.0mg,0.4mmol)和干燥且脱气的dmf(15ml),搅拌,最后加入二苯基膦(0.7g,3.9mmol)。将所得混合物在90℃搅拌6小时。冷却至室温后,加入水(20ml),产物用乙酸乙酯(15ml
×
3)萃取。合并的有机层用硫酸钠干燥并减压浓缩除去溶剂硅胶柱分离纯化,得到白色固体产物l6(1.0g,60.7%)。
[0089]
11
h nmr(400mhz,cdcl3)δ=6.86
–
6.90(m,2h),6.96
–
7.05(m,3h),7.10
–
7.20(m,6h),7.21-7.30(m,11h),7.35
–
7.41(m,4h);
31
p nmr(162mhz,cdcl3)δ=
–
27.19(d,j=144.8hz),
–
6.24(d,j=144.8hz).
[0090][0091]
(2)络合物6的制备
[0092]
在一个干燥的并充满氩气的schlenk反应管中,加入配体l6(236.1mg,0.5mmol)和crcl3(thf)3(187.3mg,0.5mmol),向其中加入重蒸二氯甲烷(10ml),室温下搅拌2h。反应结束后过滤,滤液抽干,得到固体用正己烷洗涤(5ml
×
3),抽干得到蓝色粉末203.0mg(0.42mmol,83%)。
[0093]
实施例7
[0094]
(1)配体l7的制备
[0095]
在一个充满氩气的50ml schlenk烧瓶中,加入叔丁基乙炔(0.8g,10.0mmol)和10ml重蒸四氢呋喃,搅拌并冷却至0℃,将正丁基锂(4.00ml,2.5m己烷溶液,10.0mmol)缓慢加入溶液中,在该温度下搅拌30分钟。然后逐滴加入ph2pcl(2.20g,10.0mmol),并将混合物升温至环境温度并搅拌1小时。真空除去挥发物后,混合物用石油醚(30ml)萃取。然后过滤所得混合物以除去不溶性盐,真空干燥滤液,得到白色固体产物(2.60g,95.8%)。
[0096]
在一个充满氩气的schlenk管中,加入上述白色固体产物(931.4mg,3.5mmol)、碘化亚铜(34.0mg,0.2mmol)、碳酸铯(114.0mg,0.4mmol)和干燥且脱气的dmf(15ml),搅拌,最后加入二苯基膦(0.7g,3.9mmol)。将所得混合物在90℃搅拌6小时。冷却至室温后,加入水(20ml),产物用乙酸乙酯(15ml
×
3)萃取。合并的有机层用硫酸钠干燥并减压浓缩除去溶剂硅胶柱分离纯化,得到白色固体产物l7(1.1g,67.2%)。
[0097]1h nmr(400mhz,cdcl3)δ=1.26(s,9h),6.94
–
7.07(m,12h),7.20
–
7.27(m,4h),7.31(dd,j=16.0,3.5hz,1h),7.62
–
7.68(m,4h);
31
p nmr(162mhz,cdcl3)δ=
–
30.26(d,j=29.3hz),
–
11.35(d,j=29.3hz).
[0098]
[0099]
(2)络合物7的制备
[0100]
在一个干燥的并充满氩气的schlenk反应管中,加入配体l7(226.1mg,0.5mmol)和crcl3(thf)3(187.3mg,0.5mmol),向其中加入重蒸二氯甲烷(10ml),室温下搅拌2h。反应结束后过滤,滤液抽干,得到固体用正己烷洗涤(5ml
×
3),抽干得到蓝色粉末306.5mg(0.45mmol,90.0%)。
[0101]
实施例8
[0102]
(1)催化剂的制备
[0103]
在一个干燥的并充满氩气的schlenk反应管中,加入络合物1(0.33mg,0.50μmol)、重蒸的甲基环己烷(30ml),搅拌5分钟后加入改性甲基铝氧烷mmao-3a(0.4mmol,1.12mol/l),室温反应5分钟后备用。
[0104]
(2)乙烯齐聚反应
[0105]
将200ml的不锈钢高压气体反应釜,在120℃的油浴上抽真空3小时保证反应釜的无水无氧环境,然后冷却至反应温度,用乙烯气体对釜内置换气三次。然后立即用干燥的玻璃注射器吸取上述制备好的催化剂溶液注入到高压反应釜中,密封反应釜,开启搅拌,并通入乙烯气体,调压至4.0mpa,60℃搅拌反应60分钟。反应结束后,关闭乙烯供气阀门,冷却至0℃,泄压,打开反应釜,加入定量内标壬烷并搅拌均匀。随后用10wt%的hcl水溶液大约30ml淬灭反应,取少量有机相过滤后进行gc分析。反应釜中剩下的混合物过滤后取固体,将固体加入10wt%hcl水溶液中搅拌2小时,过滤,烘干至恒重后称重,数据见表1。
[0106]
实施例9
[0107]
与实施例8不同之处在于络合物1替换为络合物2(0.30mg,0.5μmol),数据见表1。
[0108]
实施例10
[0109]
与实施例8不同之处在于所用络合物1替换为络合物3(0.31mg,0.5μmol),数据见表1。
[0110]
实施例11
[0111]
与实施例8不同之处在于所用络合物1替换为络合物4(0.28mg,0.5μmol),数据见表1。
[0112]
实施例12
[0113]
与实施例11不同之处在于乙烯齐聚反应在35℃下进行,数据见表1。
[0114]
实施例13
[0115]
与实施例11不同之处在于乙烯齐聚反应在80℃下进行,数据见表1。
[0116]
实施例14
[0117]
与实施例11不同之处在于mmao-3a用量为0.3mmol,数据见表1。
[0118]
实施例15
[0119]
与实施例11不同之处在于mmao-3a用量为0.5mmol,数据见表1。
[0120]
实施例16
[0121]
与实施例11不同之处在于乙烯齐聚的反应压力为2.0mpa,数据见表1。
[0122]
实施例17
[0123]
与实施例11不同之处在于乙烯齐聚的反应压力为1.0mpa,数据见表1。
[0124]
对比例1
[0125]
与实施例11不同之处在于络合物4替换为络合物5(0.35mg,0.5μmol),数据见表1。
[0126]
对比例2
[0127]
与实施例11不同之处在于络合物4替换为络合物6(0.35mg,0.5μmol),数据见表1。
[0128]
对比例3
[0129]
与实施例11不同之处在于络合物4替换为络合物7(0.34mg,0.5μmol),数据见表1。
[0130]
表1
[0131][0132]
从表1可以看出,本发明提供的催化剂催化活性较高,最高可达到3146kg/g cr/h,并且1-辛烯选择性达到75.7%,最高可达81.0%。与对比例1至对比例3相比,本发明提供的催化剂催化活性和1-辛烯选择性均有较大的提升,与本发明的构思相互印证。
[0133]
本发明中,配体l1~l4和三个对比的含四个苯基的配体l5~l7,l1~l4的催化活性,1-辛烯的选择性均高于l5~l7,而且含l1~l4的催化剂在反应中产生的聚合物极少(0.05%~0.1%),大幅度小于l5~l7(1.1~1.3%),与本发明的构思相互印证。
[0134]
以上所述,仅是本发明的较佳实施例而已,并非是对本发明作其它形式的限制,任何熟悉本专业的技术人员可能利用上述揭示的技术内容加以变更或改型为等同变化的等效实施例。但是凡是未脱离本发明技术方案内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与改型,仍属于本发明技术方案的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1