一种中温疏水脱碳吸附剂及其制备方法
1.本发明涉及清洁能源技术领域,特别涉及一种中温疏水脱碳吸附剂及其制备方法。
背景技术:
2.工业生产和日常生活中,二氧化碳(co2)气体的产生和排放不可避免。该类气体会造成大气层温室效应,破坏地球的生态环境。煤制氢以及烃类重整制氢工业中,需要对co2等含碳杂质气体组分进行脱除。传统溶液吸收吸附等方法在低于100℃温度区间使用,造成能源的浪费,且成本较高,且存在二次污染,因而干法脱碳在节能减排和降低成本方面具有更好的前景。
3.在干法脱碳工艺中,吸附剂的合成和应用尤其重要。其中,活性炭吸附剂具有发达的孔隙结构和巨大的比表面积,对气体、液体中的含碳杂质组分(以co2和co为主)都具有较强的吸附。然而,当活性炭用作干法脱碳的吸附剂时,由于活性炭表面的毛细管现象,其表面大量吸附环境中的水,使得在吸附过程中脱碳的能力大幅度下降;诸如煤制氢以及烃类重整制氢工业中净化前混合气温度较高(200~350
o
c)且含水蒸气。因此,合成既具有较大比表面积、较强脱碳吸附能力,又在中温条件下具有较好疏水能力的活性炭吸附剂,是此类材料开发的重要要求。
技术实现要素:
4.有鉴于此,本发明的目的在于提供一种中温疏水脱碳吸附剂及其制备方法。本发明提供的中温疏水脱碳吸附剂既具有较大比表面积、较强脱碳吸附能力,又具有较好疏水能力、在中温条件下保持较好的脱碳吸附能力。
5.本发明提供了一种中温疏水脱碳吸附剂的制备方法,包括以下步骤:a)将碳源、氮源与有机溶剂混合后干燥,得到一级前驱体;b)对所述一级前驱体进行炭化处理,得到炭化产物;c)将所述炭化产物与氯化锌、有机溶剂混合后干燥,得到二级前驱体;d)将所述二级前驱体进行热活化处理,得到中温疏水脱碳吸附剂;所述碳源为无烟煤和中温煤沥青。
6.优选的,所述氮源为尿素和/或三聚氰胺。
7.优选的,所述无烟煤的灰分≤10%;所述中温煤沥青的细度为≥300目;所述步骤a)中,所述有机溶剂为乙醇和四氢呋喃。
8.优选的,所述无烟煤在所述碳源中的质量比为70%~90%;所述碳源与氮源的质量比为(80~120)∶(80~240);所述步骤a)中,碳源与有机溶剂的质量比为(80~120)∶(200~1200);所述步骤a)中,所述干燥的温度为70~100℃,时间为4~6 h。
9.优选的,所述步骤b)中,所述炭化处理的终点温度为550~600℃,保温时间为180~
200 min。
10.优选的,所述步骤c)中:所述炭化产物与氯化锌的质量比为100∶(50~200);所述炭化产物与有机溶剂的质量比为100∶(300~400);所述有机溶剂为乙醇;所述干燥的温度为60~70℃。
11.优选的,所述步骤d)中,所述热活化处理的温度为700~900℃,保温时间为120~180 min。
12.优选的,所述步骤d)中,在所述热活化处理后,还进行酸洗处理;所述酸洗处理采用的酸液为盐酸液;所述酸液的浓度为1~2m。
13.优选的,所述炭化处理在保护性气氛中进行;所述热活化处理在保护性气氛中进行。
14.本发明还提供了一种上述技术方案中所述的制备方法制得的中温疏水脱碳吸附剂。
15.本发明制备的中温疏水脱碳吸附剂为氮掺杂疏水活性炭,其以无烟煤和中温煤沥青作为碳源,引入氮源,并以氯化锌作为活化造孔剂。制备过程中先通过有机溶剂将碳源和氮源形成粘稠交联分散体系,并通过干燥获得一级前驱体;然后,进行炭化处理,得到炭化产物;之后,再将炭化产物、氯化锌和有机溶剂形成粘稠分散体系,并通过干燥获得二级前驱体;最后再通过热活化处理,得到中温疏水脱碳吸附剂。所得吸附剂产物为粉末状,颗粒为微米级,微观尺寸极小,容易通过各类成形造粒方法加工为吸附剂成品。其氮掺杂的引入不仅形成了石墨型氮(n
‑
q)掺杂,通过原位掺杂破坏羰基化合物(降低表面氧含量、同时提升表面氮含量)提高材料疏水性。而且高温活化下尿素/三聚氰胺这两种氮源可以形成吡咯型氮(n
‑
5)和吡啶型氮(n
‑
6)。这两类氮掺杂形式可以使材料表面接触水分子时,推动水分子电离平衡向右移动,使表面显碱性,增大co2一类酸性气体在其表面吸附平衡常数,从而提升其吸附容量,以此增大材料表面对co2的吸附选择性。本发明制得的中温疏水脱碳吸附剂,除了一般干法脱除co2应用场合,尤其适用于中温区间(120~350
o
c)、从含有水蒸气组分的原料混合气体中脱除co2。
16.实验结果表明,本发明制得的中温疏水脱碳吸附剂的比表面积在65m2/g以上,接触角在120
°
以上,常温下对co2吸附量在1.8mmol/g以上;而且,在中温条件下(温度≥150℃),对co2吸附量仍在1.1mmol/g以上。
附图说明
17.为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据提供的附图获得其他的附图。
18.图1为对比例1所得样品的sem图;图2为实施例1所得样品的sem图;图3为对比例1、对比例2、实施例1和实施例2所得产品的水接触角测试结果图;
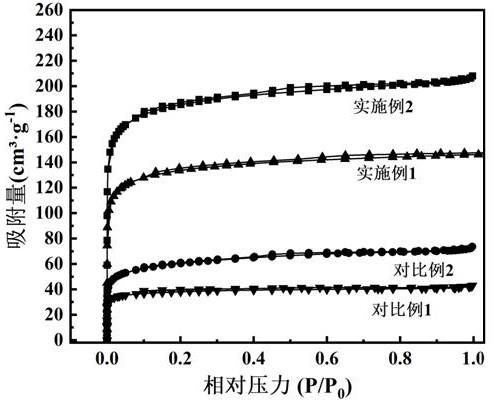
图4为实施例1、实施例2、对比例1和对比例2所得样品的bet吸附曲线图;图5为实施例1所得样品的n 1sx射线光电子能谱分析图;图6为实施例2所得样品的n 1s x射线光电子能谱分析图;图7为实施例1所得样品的x射线光电子能谱总能谱;图8为对比例1所得样品的x射线光电子能谱总能谱;图9为实施例1样品在常温和中温条件下的co2等温吸附曲线图;图10为实施例2样品在常温和中温条件下的co2等温吸附曲线图;图11为实施例1和对比例1所得样品通过热重法在150℃下的co2等温吸附曲线图;图12为实施例1样品通过固定床穿透试验、模拟测试150℃下在不同的固定床含水蒸气量co2图;图13为对比例1样品通过固定床穿透试验、模拟测试150℃下在不同的固定床含水蒸气量co2图。
具体实施方式
19.本发明提供了一种中温疏水脱碳吸附剂的制备方法,包括以下步骤:a)将碳源、氮源与有机溶剂混合后干燥,得到一级前驱体;b)对所述一级前驱体进行炭化处理,得到炭化产物;c)将所述炭化产物与氯化锌、有机溶剂混合后干燥,得到二级前驱体;d)将所述二级前驱体进行热活化处理,得到中温疏水脱碳吸附剂;所述碳源为无烟煤和中温煤沥青。
20.关于步骤a):将碳源、氮源与有机溶剂混合后干燥,得到一级前驱体。
21.本发明中,所述碳源为无烟煤和中温煤沥青。
22.所述无烟煤是碳元素分布最均衡、碳有序度最高的煤原料。无烟煤的含碳量可以达到90%,灰分和磷、硫等杂质含量总和不超过10%,这对于制备活性炭材料时炭化后的得碳率有了基本的保证。无烟煤具有较发达的孔洞,在本发明的制备体系中,方便被溶解、分散的氮源和吸附剂吸附与孔道内,使得碳源与氮源、活化剂的分散更加均匀。本发明中,所述无烟煤的粒度≥300目,优选为300~350目;若颗粒粒度过小,则无法吸附更多的氮源和活化剂,若颗粒过大则材料比表面积受限,产品吸附能力下降。
23.所述中温煤沥青(简称ctp)是指软化点在90~100℃的煤沥青。煤沥青没有具体的熔点和凝固点,为玻璃体。当其到达软化点后开始失去固态形状,在软化点100℃以上彻底成为液态。本发明中,在后续炭化过程中,ctp完全液化的温度需要与氮源受热分解并分散的中间产物匹配,方便该过程各类产物均匀分散,提高氮掺杂在材料表面的均匀度和产物的交联度。因此,本发明选择ctp和特定的氮源(尿素和/或三聚氰胺)搭配。尿素在170℃开始受热分解生成双缩脲和三聚氰酸,进而生成三缩脲;三聚氰胺受热分解温度为350℃,生成密勒胺(c6h6n
10
),因而作为碳源的一部分,本发明起到交联粘结的作用的ctp完全液化的温度须低于该温度,在氮源完全分解之前液化,从而将氮源分解产物、ctp、无烟煤均匀地混合在一起。低温煤沥青在100 ℃以下就会液化,导致前驱体溶剂挥发时会随着溶剂一起挥发;高温煤沥青到350℃也无法完全液化,会形成碳源、氮源分布不均匀,本发明使用ctp才能达到物料分散均匀、提高氮掺杂在材料表面的均匀度和产物的交联度的效果,使产物获
得最佳性能。在本发明的一些实施例中,所述ctp含碳量为73.5%,符合国标gb2209
‑
2012规格。本发明中,所述ctp的粒度≥300目,优选为300~350目。
24.本发明中,所述无烟煤占所述碳源的质量比为70%~90%,优选为85%~90%,最优选为90%。本发明通过控制两种碳源的比例,调控前驱体黏性以及成品中无定形碳与结晶碳的微观交联程度;小试研究中证明,在上述比例下,两种碳源的交联分散效果最好,炭化过程中的损失最小。
25.本发明中,所述氮源为尿素(ch4n2o,含氮量47%)和/或三聚氰胺。相比其它氮源物质,本发明采用上述特定氮源,才能使产品达到最佳的脱碳吸附效果。尿素类化合物受热分解生成双缩脲、三缩脲,进而生成g
‑
c3n4;三聚氰胺受热分解生成密勒胺(c6h6n
10
)进而生成g
‑
c3n4。这种物质g
‑
c3n4是微米级片状物,片的尺寸和厚度都很小,有利于其均匀附着在无烟煤颗粒表面以及涌入大尺寸孔道内部,使得材料碳、氮分布均匀,而不是碳与碳相连、氮与氮成团聚集。能在炭化中产生该物质的有机物很多,比如双缩脲,单氰胺,双氰胺等,但是尿素类化合物中,双缩脲和三缩脲有较大的腐蚀性和气味,比起尿素相对安全性低。而氰胺类化合物中,单氰胺和双氰胺会与不溶物颗粒形成胶核,使得前驱体各组分分散性变差,而三聚氰胺形成的g
‑
c3n4的片层厚度大概在200 nm左右,平面大小在10
×
10 μm左右,这样的尺寸有利于交联和分割,防止无烟煤基颗粒的堆叠团聚,使产品达到最佳的脱碳吸附效果。
26.本发明中,所述碳源与氮源的质量比为(80~120)∶(80~240),更优选为100∶(100~180)。在上述质量比下,产品的疏水性能和脱碳吸附效果较好。在本发明的一些实施例中,所述质量比为100∶180或100∶150。更具体的,在本发明的一些实施例中,氮源为尿素,所述质量比为100∶180;氮源为三聚氰胺,所述质量比为100∶150。
27.本发明中,所述有机溶剂优选为乙醇和四氢呋喃。其中,所述四氢呋喃在所述有机溶剂中的体积分数优选为25%~50%;若四氢呋喃比例过低,则溶解ctp时无法完全溶解,使ctp在前驱体中与其他组分混合不够均匀;若四氢呋喃比例过高则乙醇含量不够,以尿素为氮源的时候,无法充分溶解尿素;以三聚氰胺为氮源时,无法使三聚氰胺形成乳浊液。
28.本发明中,所述碳源与有机溶剂的质量比优选为(80~120)∶(200~1200);在本发明的一些实施例中,所述质量比为100∶200。
29.本发明中,所述混合后干燥的过程优选为:先于第一温度下搅拌混合并挥发溶剂,得到粘稠膏;再于第二温度下干燥,得到一级前驱体。其中,所述第一温度优选为55~70℃;所述搅拌的速率优选为100~200rpm;所述搅拌的时间没有特殊限制,直至物料体系成为粘稠膏状即可。所述干燥的温度(即第二温度)优选为70~100℃;所述干燥的时间优选为4~6h。经上述处理,形成均匀的交联分散系,得到一级前驱体。
30.关于步骤b):对所述一级前驱体进行炭化处理,得到炭化产物。
31.本发明中,所述炭化处理优选在保护性气氛中进行。本发明对提供保护性气氛的保护性气体种类没有特殊限制,为本领域技术人员熟知的常规保护性气体即可,如氮气、氦气或氩气等。本发明对所述保护性气氛的气压没有特殊限制,为常压即可,即在常压下进行炭化处理。本发明中,所述保护性气体的流量优选为0.5~0.8 ml/min。
32.本发明中,所述炭化处理的温度为550~600℃。优选的,所述炭化处理中,在升温过程中采用三段式梯度升温方式。所述三段式梯度升温方式具体优选为:先以3~6℃/min的速率升至250~350℃,再以1~3℃/min的速率升至400~450℃,最后以2~5℃/min的速率升至终
点温度450~550℃进行保温。在本发明的一些实施例中,所述三段式梯度升温方式具体优选为:先以5℃/min的速率升至300℃,再以1℃/min的速率升至400℃,最后以2℃/min的速率升至终点温度550℃进行保温。本发明中,所述炭化处理中,保温时间优选为180~200 min;在本发明的一些实施例中,保温时间为180min。
33.采用上述三段式梯度升温方式,主要是由于在不同的温度区间内,碳源和氮源的热分解速率随着升温速率的变化是不同的,0~300 ℃内,氮源和无烟煤不进行热分解,ctp在150 ℃开始软化,到300 ℃彻底转化为液态,因此控制该温度区间内升温速率较快一点;在300~400 ℃温度区间内,尿素会转变为双缩脲和三缩脲,而三聚氰胺会转化为密勒胺,均呈现液态,若升温速率过快的话,该分解产物会加快气化离开体系,因而控制该区间升温速率较慢,保持在1 ℃/min左右。在400~550℃过程中,ctp会被彻底炭化,形成薄片状附着于无烟煤颗粒表面,而尿素热解产生的双缩脲、三缩脲,或者三聚氰胺热解产生的密勒胺,都会转变为g
‑
c3n4,也呈现碎片状,厚度约为100 nm,若快速直升至550 ℃,ctp和两种氮源的分解产物都会因为气化被保护气体大量带走,使得产品含氮量急剧降低,ctp也无法形成交联的碳碎片,影响产品的性能。因此,本发明控制在上述三段式梯度升温方式下,有利于使产品性能达到最佳。
34.经上述炭化处理,氮源形成g
‑
c3n4微米级片状物,且其均匀附着在碳源颗粒表面以及涌入大尺寸孔道内部,获得碳、氮分布均匀的炭化产物。
35.关于步骤c):将所述炭化产物与氯化锌、有机溶剂混合后干燥,得到二级前驱体。
36.本发明中,采用氯化锌作为活化造孔剂,利用高温活化条件下碳元素还原锌元素进行造孔,获得具有良好疏水性和高比表面积的活性炭材料。本发明中,所述炭化产物与氯化锌的质量比优选为100∶(50~200);在本发明的一些实施例中,所述质量比为100∶100或100∶200。
37.本发明中,所述有机溶剂优选为乙醇。本发明中,所述炭化产物与有机溶剂的质量比优选为100∶(300~400)。
38.本发明中,所述混合后干燥的过程优选为:先于第一温度下搅拌混合并挥发溶剂;再于第二温度下干燥,得到二级前驱体。其中,所述第一温度优选为55~65℃;所述搅拌的速率优选为100~200rpm;所述搅拌的时间没有特殊限制,直至物料体系中溶剂挥发使物料成为粘稠膏状即可。所述干燥的温度(即第二温度)优选为60~70℃。经上述处理,得到二级前驱体。
39.关于步骤d):将所述二级前驱体进行热活化处理,得到中温疏水脱碳吸附剂。
40.本发明中,所述热活化处理优选在保护性气氛中进行。本发明对提供保护性气氛的保护性气体种类没有特殊限制,为本领域技术人员熟知的常规保护性气体即可,如氮气、氦气或氩气等。本发明对所述保护性气氛的气压没有特殊限制,为常压即可,即在常压下进行热活化处理。本发明中,所述保护性气体的流量优选为0.5~0.8 ml/min。
41.本发明中,所述热活化处理的温度为700~900℃;所述热活化处理的保温时间为120~180 min。本发明中,所述热活化处理中,在升温过程中优选采用两段式梯度升温方式。所述两段式梯度升温方式具体优选为:先以3~6℃/min的速率升至500~560℃,再以2~4℃/min的速率升至终点温度700~900℃进行保温。在本发明的一些实施例中,所述两段式梯度升温方式具体优选为:先以5℃/min的速率升至550℃,再以2℃/min的速率升至700~900℃
进行保温。在本发明的一些实施例中,所述热活化处理的终点保温温度为700℃、800℃或900℃,更优选为700~800℃。
42.本发明采用上两段式梯度升温方式,先以较快速率升温,再降低速率升温;在550 ℃后,氯化锌会被碳元素还原成zn单质,如果升温过快的话,氯化锌晶体会大量地直接逸散,被保护气氛带走,起不到活化的作用,因而第二段须降低升温速率。通过上述处理,能够达到最佳的活化效果。
43.本发明中,经上述热活化处理后,优选还进行酸洗处理。所述酸洗处理采用的酸液优选为盐酸液。所述盐酸液的浓度优选为1~2m。所述酸洗的时间优选为6~8h。经酸洗后,优选还进行水洗和干燥。所述干燥的温度优选为60~90℃。经以上后处理,得到氮掺杂疏水活性炭这种中温疏水脱碳吸附剂。
44.本发明中,所述中温疏水脱碳吸附剂记为u
z
a
x
c
y
z
m
‑
t或m
z
a
x
c
y
z
m
‑
t,其中,u和m代表氮源,a代表无烟煤,c代表中温煤沥青,z代表氯化锌活化剂,t代表热活化处理的温度。
45.本发明提供的上述制备方法,将无烟煤与一定比例的ctp作为碳源,加入不同种类和比例的氮源,以氯化锌为活化剂,通过中温炭化、高温活化,利用高温条件下碳元素还原锌元素进行造孔,获得具有良好疏水性和高比表面积的活性炭材料。原料经中温炭化、高温活化后呈粉末状,且具有较大的比表面积和较强的机械硬度,同时在中温条件下具有较好的疏水性能(水接触角>120
°
),在提高其对于含碳气体的吸附量的同时,尽可能降低水蒸气对于吸附剂碳容量的负面影响。上述工艺简单,原材料成本低廉。作为碳源的煤沥青为煤焦化工业的副产物,三聚氰胺和氯化锌也是常用的工业品,产品易转向工业化批量生产;吸附剂产物为粉末状,颗粒为微米级,微观尺寸极小,容易通过各类成形造粒方法加工为吸附剂成品。其氮掺杂的引入不仅形成了石墨型氮掺杂,通过原位掺杂破坏羰基化合物提高材料疏水性,而且使得材料表面遇水呈碱性,使得吸附co2等酸性气体的平衡进一步向右移动,提升其吸附容量;且结构稳定,在中温条件下具有较好疏水能力、保持较好的脱碳吸附能力。
46.本发明还提供了一种上述技术方案中所述的制备方法制得的中温疏水脱碳吸附剂。上述中温疏水脱碳吸附剂可用作多组分含水气体中co2脱除以及其他含碳气体脱除的吸附剂;除了一般干法脱除co2应用场合,尤其适用于中温区间(120~350
o
c)、从含有水蒸气组分的原料混合气体中脱除co2。
47.实验结果表明,本发明制得的中温疏水脱碳吸附剂的比表面积在65m2/g以上,接触角在120
°
以上,常温下对co2吸附量在1.8mmol/g以上;而且,在中温条件下(温度≥150℃),对co2吸附量仍在1.1mmol/g以上。
48.为了进一步理解本发明,下面结合实施例对本发明优选实施方案进行描述,但是应当理解,这些描述只是为进一步说明本发明的特征和优点,而不是对本发明权利要求的限制。
49.以下各实施例和对比例中,无烟煤购自国家投资集团神华宁夏煤业集团有限责任公司。ctp购自邯郸市延金贸易有限公司,含碳量为73.5wt%。以下实施例中,所得中温疏水脱碳吸附剂记为u
z
a
x
c
y
z
m
‑
t或m
z
a
x
c
y
z
m
‑
t,其中,u和m代表氮源,a代表无烟煤,c代表中温煤沥青,z代表氯化锌活化剂,t代表热活化处理的温度。实施例1~6及对比例1~3所得产物对应的标记代号参见表1。
50.表1 实施例及对比例产物对应的标记代号实施例1:制备u
15
a9c1z
10
‑
700氮掺杂疏水活性炭s1、制备一级前驱体称取10份ctp溶于200份四氢呋喃
‑
乙醇混合溶剂中(四氢呋喃在混合溶剂中的体积分数为25%),再称取180份尿素,溶解于上述溶剂中。之后,称取90份无烟煤,分散于上述体系中,恒温水浴加热至65℃,以150 rpm的转速搅拌混匀同时挥发溶剂,待成为粘稠膏状物时(此时加热搅拌了60min),转移至氧化铝石英舟中,于70℃鼓风干燥5h,得到一级前驱体,记为[a]9[c]1[u]
15
。
[0051]
其中,无烟煤含碳量计为90 %,ctp含碳量记为75 %。尿素含氮量记为52 %。要求前驱体内碳氮比达到10:15,则100份该比例碳源,对应180份尿素。因此,所得一级前驱体计为[a]9[c]1[u]
15
。
[0052]
s2、炭化处理将所得一级前驱体[a]9[c]1[u]
15
置于管式炉中,通入流量为0.6 ml/min的高纯氮气作为保护气体,以5℃/min的速率升温至300℃,再以1℃/min的速率升温至400℃,最后以2℃/min的速率升温至550℃,保温3h。随炉冷却至室温后取出,用1000份去离子水浸泡清洗、抽滤并于70℃鼓风干燥2h,得到炭化产物。
[0053]
s3、制备二级前驱体称取100份氯化锌,溶解于350份乙醇中,再称取100份炭化产物分散于体系中,恒温水浴加热至60℃,以150 rpm的转速搅拌混匀同时挥发溶剂,待成为粘稠膏状物时(此时加热搅拌了60min),转移至氧化铝石英舟中,于70℃鼓风干燥箱内完全干燥,得到二级前驱体,记为[a]9[c]1[u]
15
[zn]
10
。
[0054]
s4、热活化处理将二级前驱体[a]9[c]1[u]
15
[zn]
10
平放于管式炉的恒温区中,通入流量为0.6 ml/min的高纯氮气作为保护气体,以5℃/min的速率升温至550℃,再以2℃/min的速率升温至700℃,保温2h。随炉冷却至室温后取出,用浓度1.5m的盐酸液酸洗7h后,用总计1200质量份的去离子水清洗抽滤3次,再于70℃鼓风干燥完全,得到氮掺杂活性炭,记为u
15
a9c1zn
10
‑
700。
[0055]
实施例2:制备m
15
a9c1z
10
‑
700氮掺杂疏水活性炭1、样品的制备:s1、制备一级前驱体称取10份ctp溶于200份四氢呋喃
‑
乙醇混合溶剂中(四氢呋喃在混合溶剂中的体积分数为25%),再称取150份三聚氰胺,以6份/min的速率将三聚氰胺加入分散体系中,直至形成不沉降的乳浊液分散体系。将分散体系于恒温水浴加热至65℃,以150 rpm的转速搅拌混匀同时挥发溶剂,待成为粘稠膏状物时,转移至氧化铝石英舟中,于100℃鼓风干燥5h,得到一级前驱体,记为[a]9[c]1[m]
15
。
[0056]
s2、炭化处理将所得一级前驱体[a]9[c]1[m]
15
置于管式炉中,通入流量为0.6 ml/min的高纯氮气作为保护气体,以5℃/min的速率升温至300℃,再以1℃/min的速率升温至400℃,最后以2℃/min的速率升温至550℃,保温3h。随炉冷却至室温后取出,用1000份去离子水浸泡清洗、抽滤并于70℃鼓风干燥2h,得到炭化产物。
[0057]
s3、制备二级前驱体称取100份氯化锌,溶解于350份乙醇中,再称取100份炭化产物分散于体系中,恒温水浴加热至65℃,以150 rpm的转速搅拌混匀同时挥发溶剂,待成为粘稠膏状物时,转移至氧化铝石英舟中,于70℃鼓风干燥箱内完全干燥,得到二级前驱体,记为[a]9[c]1[m]
15
[zn]
10
。
[0058]
s4、热活化处理将二级前驱体[a]9[c]1[m]
15
[zn]
10
平放于管式炉的恒温区中,通入流量为0.6 ml/min的高纯氮气作为保护气体,以5℃/min的速率升温至550℃,再以2℃/min的速率升温至700℃,保温2h。随炉冷却至室温后取出,用浓度1.5m的盐酸液酸洗7h后,用总计1200质量份的去离子水清洗抽滤3次,再于70℃鼓风干燥完全,得到氮掺杂活性炭,记为m
15
a9c1zn
10
‑
700。
[0059]
对比例1:制备a9c1z
10
‑
700氮掺杂疏水活性炭按照实施例1的制备过程实施,不同的是,步骤s1中不添加氮源尿素。
[0060]
对比例2:制备u
15
a
10
zn
10
‑
700氮掺杂疏水活性炭按照实施例1的制备过程实施,不同的是,将步骤s1中的ctp替换为等量的无烟煤(即碳源全部为无烟煤)。
[0061]
对比例3:制备m
15
a
10
zn
10
‑
700氮掺杂疏水活性炭按照实施例2的制备过程实施,不同的是,将步骤s1中的ctp替换为等量的无烟煤(即碳源全部为无烟煤),并将步骤s1中的尿素替换为等质量的三聚氰胺。
[0062]
上述实施例1~2及对比例1~3的样品表征、性能测试及分析:1、sem表征对实施例1和对比例1所得样品进行微观形貌表征,分别参见图1和图2,图1为对比例1所得样品的sem图,图2为实施例1所得样品的sem图。可以看出,对比例1颗粒分布不均匀,呈现严重的团聚现象,而且表面孔的分布不均匀。而实施例1由于加入了尿素作为氮源,在高温下无烟煤崩解为较小颗粒,并与碳源形成的碎片彼此交联,结构均匀,大孔、介孔和微孔彼此形成孔道,避免了碳源颗粒之间的团聚和坍缩导致的界面收缩。
吸附量为1.81mmol/g,150℃测试条件下co2吸附量为1.201 mmol/g;实施例2样品在30℃测试条件下的co2吸附量为2.104mmol/g,150℃测试条件下co2吸附量为1.316 mmol/g。
[0070]
(2)实施例1和对比例1样品在中温条件下对co2的吸附性测试通过热重法在150℃下测试实施例1样品的co2等温吸附线,并与实施例1的效果进行对比,结果如图11所示,图11为实施例1和对比例1所得样品通过热重法在150℃下的co2等温吸附曲线图。可以看出,在中温条件下,实施例1样品均对co2的吸附性明显高于对比例1样品,具体的,实施例1样品的co2吸附量为1.201 mmol/g,而对比例1样品的co2吸附量为0.605 mmol/g。同时,同样对对比例2样品进行测试,结果显示,对比例2样品的co2吸附量为0.852 mmol/g。对比例2中不添加ctp,使得一级前驱体未形成交联状,导致其炭化过程中前驱体被气流吹散较多,并且氮源无法均匀地渗入碳源内,导致其水接触角较小,并且co2吸附能力也较差。
[0071]
同样的,对对比例3的样品也进行以上测试。实施例1~2及对比例1~3所得产品的各项表征测试效果对比参见表2。
[0072]
表2 实施例1
‑
2产品和对比例1
‑
3产品的效果6、基于固定床突破实验的蒸气耐受性测试:各个样品对蒸气的耐受性及其同时对co2的吸附性能通过固定床反应器测试。在测试过程中,将尺寸为0.5~5 mm的120 g活性炭样品以约10 cm的填充高度放置在固定床反应器中,固定床温度稳定在200℃,进口原料气(co2或he,1个大气压)和蒸气的流量分别由质量流量计和恒流量泵调节,并使用质谱仪在线检测出口气体成分。测试前,将活性炭用he吹扫24 h(50 ml/min)以确保其表面没有吸附其他分子。随后,将吹扫气体改变为he/水蒸气混合气体,并将总流速设定为200 ml/min。通过调节氦气和水蒸气的流量比,使吹扫气体的湿度变化,变化范围为干燥(0%水蒸气)到纯水蒸气(100%水蒸气)。经过60 min的湿气吹扫后,固定床中的气体湿度达到稳定。随后将原料气更改为co2(50 ml/min),并记录穿透时间。每次测试后,吸附剂均使用he完全再生。彻底再生的标准是出口气体中的co2浓度低于10 ppm(气体含量的变化过程由在线气体分析仪进行检测)。
[0073]
实施例1和对比例1样品的测试结果分别参见图12和图13,图12为实施例1样品通过固定床穿透试验、模拟测试150℃下在不同的固定床含水蒸气量co2图,图13为对比例1样
品通过固定床穿透试验、模拟测试150℃下在不同的固定床含水蒸气量co2图。可以明显看出,随着湿度的增加,对比例1的co2吸附性能下降。将co2突破的标准设定为出口co2浓度为20%,在不同条件下的突破时间分别约为295 s(水蒸气含量0%),300 s(10%),270 s(50%),210 s(66%)和30 s(100%)。对于实施例1,在水蒸气百分比为0%、10%、50%、66%和100%时,穿透时间分别为195s、185s、145s、175s和150 s。由此可知,在湿度较高的条件下,实施例1性能优于对比例1。而且当用纯水蒸气吹扫实施例1时(图12
‑
13中100%对应工况),吸附剂表现出稳定的co2吸附能力,而对比例1的脱碳性能随湿度的增加而明显变差。
[0074]
实施例3:制备u
15
a9c1z
10
‑
800氮掺杂疏水活性炭1、样品的制备:s1、制备一级前驱体称取10份ctp溶于200份四氢呋喃
‑
乙醇混合溶剂中(四氢呋喃在混合溶剂中的体积分数为25%),再称取180份尿素,溶解于上述溶剂中。之后,称取90份无烟煤,分散于上述体系中,恒温水浴加热至70℃,以150 rpm的转速搅拌混匀同时挥发溶剂,待成为粘稠膏状物时,转移至氧化铝石英舟中,于100℃鼓风干燥5h,得到一级前驱体,记为[a]9[c]1[u]
15
。
[0075]
s2、炭化处理将所得一级前驱体[a]9[c]1[u]
15
置于管式炉中,通入流量为0.6 ml/min的高纯氮气作为保护气体,以5℃/min的速率升温至300℃,再以1℃/min的速率升温至400℃,最后以2℃/min的速率升温至550℃,保温3h。随炉冷却至室温后取出,用1000份去离子水浸泡清洗、抽滤并于70℃鼓风干燥2h,得到炭化产物。
[0076]
s3、制备二级前驱体称取100份氯化锌,溶解于350份乙醇中,再称取100份炭化产物分散于体系中,恒温水浴加热至60℃,以150 rpm的转速搅拌混匀同时挥发溶剂,待成为粘稠膏状物时,转移至氧化铝石英舟中,于70℃鼓风干燥箱内完全干燥,得到二级前驱体,记为[a]9[c]1[u]
15
[zn]
10
。
[0077]
s4、热活化处理将二级前驱体[a]9[c]1[u]
15
[zn]
10
平放于管式炉的恒温区中,通入流量为0.6 ml/min的高纯氮气作为保护气体,以5℃/min的速率升温至550℃,再以2℃/min的速率升温至800℃,保温2h。随炉冷却至室温后取出,用浓度1.5m的盐酸液酸洗7h后,用总计1200质量份的去离子水清洗抽滤3次,再于70℃鼓风干燥完全,得到氮掺杂活性炭,记为u
15
a9c1zn
10
‑
800。
[0078]
2、样品的测试:按照前文所述的测试方法对实施例4所得样品进行测试,结果显示:样品的比表面积为192 m2/g;水接触角为128
°
;对所得样品进行x射线光电子能谱分析,结果显示,其氮掺杂总量为4.9wt%,所有氮中,对疏水性有效提升的石墨型氮,占总体氮的19.8wt%。通过热重法在150℃下测试co2等温吸附曲线,计算得到co2吸附量为1.256 mmol/g。
[0079]
实施例4:制备m
15
a9c1z
10
‑
800氮掺杂疏水活性炭1、样品的制备:s1、制备一级前驱体称取10份ctp溶于200份四氢呋喃
‑
乙醇混合溶剂中(四氢呋喃在混合溶剂中的体
积分数为25%),称取90份无烟煤,分散于上述溶液中。将上述溶液置于超声震荡中,称取150份三聚氰胺,以6份/min的速率将三聚氰胺加入分散体系中,直至形成不沉降的乳浊液分散体系。将分散体系于恒温水浴加热至65℃,以150 rpm的转速搅拌混匀同时挥发溶剂,待成为粘稠膏状物时,转移至氧化铝石英舟中,于100℃鼓风干燥5h,得到一级前驱体,记为[a]9[c]1[m]
15
。
[0080]
s2、炭化处理将所得一级前驱体[a]9[c]1[m]
15
置于管式炉中,通入流量为0.6 ml/min的高纯氮气作为保护气体,以5℃/min的速率升温至300℃,再以1℃/min的速率升温至400℃,最后以2℃/min的速率升温至550℃,保温3h。随炉冷却至室温后取出,用1000份去离子水浸泡清洗、抽滤并于70℃鼓风干燥2h,得到炭化产物。
[0081]
s3、制备二级前驱体称取100份氯化锌,溶解于350份乙醇中,再称取100份炭化产物分散于体系中,恒温水浴加热至65℃,以150 rpm的转速搅拌混匀同时挥发溶剂,待成为粘稠膏状物时,转移至氧化铝石英舟中,于70℃鼓风干燥箱内完全干燥,得到二级前驱体,记为[a]9[c]1[m]
15
[zn]
10
。
[0082]
s4、热活化处理将二级前驱体[a]9[c]1[m]
15
[zn]
10
平放于管式炉的恒温区中,通入流量为0.6 ml/min的高纯氮气作为保护气体,以5℃/min的速率升温至550℃,再以2℃/min的速率升温至800℃,保温2h。随炉冷却至室温后取出,用浓度1.5m的盐酸液酸洗7h后,用总计1200质量份的去离子水清洗抽滤3次,再于70℃鼓风干燥完全,得到氮掺杂活性炭,记为m
15
a9c1zn
10
‑
800。
[0083]
2、样品的测试:按照前文所述的测试方法对实施例5所得样品进行测试,结果显示:样品的比表面积为169 m2/g;水接触角为106
°
;通过热重法在150℃下测试co2等温吸附曲线,计算得到co2吸附量为1.263 mmol/g。
[0084]
实施例5:制备u
15
a9c1z
20
‑
800氮掺杂疏水活性炭1、样品的制备:s1、制备一级前驱体称取10份ctp溶于200份四氢呋喃
‑
乙醇混合溶剂中(四氢呋喃在混合溶剂中的体积分数为25%),再称取180份尿素,溶解于上述溶剂中。之后,称取90份无烟煤,分散于上述体系中,恒温水浴加热至65℃,以150 rpm的转速搅拌混匀同时挥发溶剂,待成为粘稠膏状物时(此时加热搅拌了60min),转移至氧化铝石英舟中,于100℃鼓风干燥5h,得到一级前驱体,记为[a]9[c]1[u]
15
。
[0085]
s2、炭化处理将所得一级前驱体[a]9[c]1[u]
15
置于管式炉中,通入流量为0.6 ml/min的高纯氮气作为保护气体,以5℃/min的速率升温至300℃,再以1℃/min的速率升温至400℃,最后以2℃/min的速率升温至550℃,保温3h。随炉冷却至室温后取出,用1000份去离子水浸泡清洗、抽滤并于70℃鼓风干燥2h,得到炭化产物。
[0086]
s3、制备二级前驱体
称取200份氯化锌,溶解于350份乙醇中,再称取100份炭化产物分散于体系中,恒温水浴加热至65℃,以150 rpm的转速搅拌混匀同时挥发溶剂,待成为粘稠膏状物时,转移至氧化铝石英舟中,于70℃鼓风干燥箱内完全干燥,得到二级前驱体,记为[a]9[c]1[u]
15
[zn]
20
。
[0087]
s4、热活化处理将二级前驱体[a]9[c]1[u]
15
[zn]
20
平放于管式炉的恒温区中,通入流量为0.6 ml/min的高纯氮气作为保护气体,以5℃/min的速率升温至550℃,再以2℃/min的速率升温至800℃,保温2h。随炉冷却至室温后取出,用浓度1.5m的盐酸液酸洗7h后,用总计1200质量份的去离子水清洗抽滤3次,再于70℃鼓风干燥完全,得到氮掺杂活性炭,记为u
15
a9c1zn
20
‑
900。
[0088]
2、样品的测试:按照前文所述测试方法对实施例6所得样品进行测试,结果显示:样品的比表面积为476 m2/g;水接触角为126
°
;对所得样品进行x射线光电子能谱分析,结果显示,其氮掺杂总量为4.9wt%,所有氮中,对疏水性有效提升的石墨型氮,占总体氮的20.2wt%。通过热重法在150℃下测试co2等温吸附曲线,计算得到co2吸附量为2.7 mmol/g。
[0089]
实施例6:制备m
15
a9c1z
20
‑
800氮掺杂疏水活性炭1、样品的制备:s1、制备一级前驱体称取10份ctp溶于200份四氢呋喃
‑
乙醇混合溶剂中(四氢呋喃在混合溶剂中的体积分数为25%),称取90份无烟煤,分散于上述溶液中。将上述溶液置于超声震荡中,称取150份三聚氰胺,以6份/min的速率将三聚氰胺加入分散体系中,直至形成不沉降的乳浊液分散体系。将分散体系于恒温水浴加热至65℃,以150 rpm的转速搅拌混匀同时挥发溶剂,待成为粘稠膏状物时,转移至氧化铝石英舟中,于100℃鼓风干燥5h,得到一级前驱体,记为[a]9[c]1[m]
15
。
[0090]
s2、炭化处理将所得一级前驱体[a]9[c]1[m]
15
置于管式炉中,通入流量为0.6 ml/min的高纯氮气作为保护气体,以5℃/min的速率升温至300℃,再以1℃/min的速率升温至400℃,最后以2℃/min的速率升温至550℃,保温3h。随炉冷却至室温后取出,用1000份去离子水浸泡清洗、抽滤并于70℃鼓风干燥2h,得到炭化产物。
[0091]
s3、制备二级前驱体称取200份氯化锌,溶解于350份乙醇中,再称取100份炭化产物分散于体系中,恒温水浴加热至65℃,以150 rpm的转速搅拌混匀同时挥发溶剂,待成为粘稠膏状物时,转移至氧化铝石英舟中,于70℃鼓风干燥箱内完全干燥,得到二级前驱体,记为[a]9[c]1[m]
15
[zn]
20
。
[0092]
s4、热活化处理将二级前驱体[a]9[c]1[m]
15
[zn]
20
平放于管式炉的恒温区中,通入流量为0.6 ml/min的高纯氮气作为保护气体,以5℃/min的速率升温至550℃,再以2℃/min的速率升温至800℃,保温2h。随炉冷却至室温后取出,用浓度1.5m的盐酸液酸洗7h后,用总计1200质量份的去离子水清洗抽滤3次,再于70℃鼓风干燥完全,得到氮掺杂活性炭,记为m
15
a9c1zn
10
‑
800。
[0093]
2、样品的测试:按照前文所述的测试方法对实施例5所得样品进行测试,结果显示:样品的比表面积为452 m2/g;水接触角为128
°
;对所得样品进行x射线光电子能谱分析,结果显示,其氮掺杂总量为4.2wt%,所有氮中,对疏水性有效提升的石墨型氮,占总体氮的23.6wt%。通过热重法在150℃下测试co2等温吸附曲线,计算得到co2吸附量为2.6mmol/g。
[0094]
由实施例5~6可知,同样前驱体碳氮比的前提下,活化剂与炭化产物的质量比为2:1时,所得样品的吸附量和孔径分布明显好于质量比为1:1时的样品。
[0095]
本文中应用了具体个例对本发明的原理及实施方式进行了阐述,以上实施例的说明只是用于帮助理解本发明的方法及其核心思想,包括最佳方式,并且也使得本领域的任何技术人员都能够实践本发明,包括制造和使用任何装置或系统,和实施任何结合的方法。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围内。本发明专利保护的范围通过权利要求来限定,并可包括本领域技术人员能够想到的其他实施例。如果这些其他实施例具有近似于权利要求文字表述的结构要素,或者如果它们包括与权利要求的文字表述无实质差异的等同结构要素,那么这些其他实施例也应包含在权利要求的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1