一种用于脱保护基的钯炭催化剂的制备方法与流程
1.本发明属于催化剂制备技术领域,具体涉及一种用于脱保护基的钯炭催化剂的制备方法。
背景技术:
2.在药物合成反应中,许多官能团存在于同一个药物分子中,在反应过程中我们只需要特定的在某些基团或者位置上发生反应,但是所选合成条件会使某些不需要参与反应的基团发生反应,从而产生副产物,甚至得不到目标产物。为此需要对相关的官能团进行保护。为使基团受到保护,而在该基团上引入的基团称为保护基团。如苄基类可作为氨基的保护基团等。而合成反应最终还需要对引入的保护基团脱除。
3.碳青霉烯类抗生素主要有:美罗培南,亚胺培南,帕尼培南,比阿培南,厄他培南,法罗培南,多尼培南等。这些培南类药物的合成反应中都需要引入pnb(对硝基苄基)和pnz(对硝基苄氧羰基)等卞基类保护基团。在其化学合成中的最后一步,需要用到钯炭催化剂进行氢化脱保护基反应,而钯炭催化剂脱保护基效率将直接影响了药物的收率。
4.专利cn102133527a提到了一种用于美罗培南合成的钯锡炭催化剂及其制备方法。该催化剂为以粉状活性炭为载体,负载活性组分金属钯和活性组分锡。制备过程经活性炭酸处理、钯和锡化合物负载得催化剂前体,催化剂前体经陈化后还原得催化剂产品。该催化剂制备工艺的特点是利用亚锡的弱还原性制备一定粒径范围的金属钯胶体,然后负载于活性炭上,但是该工艺对操作的环境温度要求苛刻,限制在0℃~5℃,给工业化生产造成了一定的不便。
5.专利cn103041805a涉及到一种培南类抗生素合成用的高活性钯炭催化剂的制备方法。该催化剂以氯亚钯酸及其盐为活性钯的前驱化合物,以粉状活性炭为载体,钯浸渍液中添加柠檬酸钠等添加剂,然后将钯浸渍液分段吸附于活性炭上,经湿化学还原而得到的钯炭催化剂。该催化剂贵金属钯含量较高增加了催化剂的使用成本。
6.专利cn103894190a涉及到一种用于美罗培南合成的钯炭催化剂的制备方法。该方法采用低含量的碱性化合物处理载体,用氯化钯盐酸溶解通过调节溶液的ph值,制备出一定大小的螯合钯离子,钯的负载量为钯炭催化剂质量的3.5%~4.5%,经化学还原而得到的钯炭催化剂。该催化剂用碱性化合物处理改性活性炭载体不利于钯均匀分散,影响了催化剂的活性。
7.目前市场上生产培南类药物所使用的钯炭催化剂存在着制造工艺复杂、脱保护基效率差、副反应多的情况,直接影响了培南类药物的收率。
技术实现要素:
8.本发明所要解决的技术问题是现有技术中存在的脱保护基效率差问题。提供一种新的用于脱保护基的钯炭催化剂的制备方法。该方法具有制造工艺简单、利于工业放大生产和推广,脱保护基效率高、产品收率高的特点。
9.为解决上述技术问题,本发明采用的技术方案如下:
10.一种用于脱保护基的钯炭催化剂的制备方法,采用改性活性炭载体,与酸性活性钯浸渍液混合后,经陈化反应再经还原反应得到所述钯炭催化剂。
11.作为本发明优选的技术方案,所述钯炭催化剂中钯负载量为所述钯炭催化剂的4.5%~5.5%。
12.作为本发明优选的技术方案,所述改性活性炭载体的制备方法包括以下步骤:将活性炭置于质量浓度为1%~10%的盐酸水溶液中沸腾回流处理1h~3h,再用去离子水洗涤至洗涤液的ph值为7~8,干燥得到第一干燥的活性炭,再将所述第一干燥的活性炭置入质量浓度为1%~10%的双氧水溶液中沸腾回流处理1h~3h,然后用去离子水洗涤至洗涤液的ph值为7~8,干燥得到第二干燥的活性炭。
13.作为本发明优选的技术方案,所述改性活性炭载体的制备方法还包括以下步骤:将所述第二干燥的活性炭用去离子水打浆,搅拌条件下稳定0.5~1h,得到所述改性活性炭载体。
14.作为本发明优选的技术方案,打浆用的所述去离子水的用量为每克所述改性活性炭载体用5ml~10ml。
15.作为本发明优选的技术方案,所述活性炭为木质活性炭,粒度为200~400目,比表面积为1200m2/g~1800m2/g。
16.作为本发明优选的技术方案,所述活性钯浸渍液的配制方法:将金属钯用王水溶解,然后加去离子水稀释后在用盐酸调节ph得到ph值为0.1~1.0所述活性钯浸渍液。
17.作为本发明优选的技术方案,所述活性钯浸渍液中钯的质量浓度为1%~5%。
18.作为本发明优选的技术方案,所述陈化反应包括所述活性钯浸渍液和所述改性活性炭载体混合之后搅拌3h~6h,再陈化处理为6h~12h。
19.作为本发明优选的技术方案,所述还原反应包括调节混合液ph值为7~8后,用水合肼还原得到所述钯炭催化剂。
20.与现有技术相比,本发明的有益效果为:
21.采用本发明的制备方法制备的钯炭催化剂,脱保护基效率跟相关技术相比有了显著的提高。本发明制造工艺简单、副反应少。
具体实施方式
22.本发明方法包括将制备改性活性炭载体、制备活性钯浸渍液以形成反应混合物的步骤。下面将更详细地描述这些组分制备的每一种。这两种产品的制备顺序是不分先后,对本领域普通技术人员来说是显而易见的。
23.一种用于脱保护基的钯炭催化剂的制备方法。采用改性活性炭载体,与酸性活性钯浸渍液混合后,经陈化反应再经还原反应得到所述钯炭催化剂。该钯炭催化剂中的钯微晶纳米颗粒状均匀分散在改性活性炭载体。
24.具体的,所述钯炭催化剂中钯负载量为所述钯炭催化剂的4.5%~5.5%。钯负载量为钯为质量占钯和活性炭的质量之和的百分数,将负载量设在此范围内,符合行业使用要求。
25.进一步的,所述改性活性炭载体的制备方法包括以下步骤:将活性炭置于质量浓
度为1%~10%的盐酸水溶液中沸腾回流处理1h~3h,再用去离子水洗涤至洗涤液的ph值为7~8,干燥得到第一干燥的活性炭,再将所述第一干燥的活性炭置入质量浓度为1%~10%的双氧水溶液中沸腾回流处理1h~3h,然后用去离子水洗涤至洗涤液的ph值为7~8,干燥得到第二干燥的活性炭。
26.另外,所述改性活性炭载体的制备方法还包括以下步骤:将所述第二干燥的活性炭用去离子水打浆,搅拌条件下稳定0.5~1h,得到所述改性活性炭载体。
27.具体的,打浆用的所述去离子水的用量为每克所述改性活性炭载体用5ml~10ml。
28.具体的,所述活性炭为木质活性炭,粒度为200~400目,比表面积为1200m2/g~1800m2/g。
29.活性钯浸渍液的配制方法:将金属钯用王水溶解,然后加去离子水稀释后在用盐酸调节ph得到ph值为0.1~1.0所述活性钯浸渍液。
30.具体的,所述活性钯浸渍液中钯的质量浓度为1%~5%。
31.另外,所述活性钯浸渍液和所述改性活性炭载体混合之后还原反应之前还包括制备催化剂前体和所述催化剂前体陈化处理,所述催化剂前体由所述活性钯浸渍液和所述改性活性炭载体混合之后搅拌3h~6h制得,所述陈化处理时间为6h~12h。
32.进一步的,所述还原反应包括调节混合液ph值为7~8后,用水合肼还原得到所述钯炭催化剂。
33.本发明先后进行多次试验,现举一部分试验结果作为参考对发明进行进一步详细描述,下面结合具体实施例进行详细说明。
34.实施例1
35.称取50克200~400目木质活性炭,比表面为1500m2/g,放入在烧瓶中。用浓度为1%wt的盐酸溶液,沸腾回流处理2h,然后用去离子水洗涤处理后的活性炭至洗涤液的ph值为7~8,烘干。再用10%wt的双氧水溶液同样处理上述炭,沸腾回流处理2h,然后用去离子水洗涤处理后的活性炭至洗涤液的ph值为7~8。烘干得到改性活性炭载体。
36.称取催化剂质量4.5%的钯2.25g,用王水溶解。用盐酸调节活性钯浸渍液的ph值为0.1,得到活性钯浸渍液。
37.上述改性活性炭载体用去离子水打浆,搅拌条件下稳定1h,得到活性炭浆液。去离子水用量为每克改性活性炭载体用10ml去离子水。将调节ph值后的活性组分活性钯浸渍液加入活性炭浆液中,搅拌3h,得到催化剂前体。
38.上述催化剂前体陈化12h后用10%wt的氢氧化钠溶液调到ph7~8,加水合肼还原。洗涤、干燥,得到4.5%wt钯炭催化剂。
39.实施例2
40.称取实施例1中相同活性炭50克。放入在烧瓶中。用浓度为5%wt的盐酸溶液,沸腾回流处理2h,然后用纯水洗涤处理后的活性炭至洗涤液的ph值为7~8,烘干。再用5%wt的双氧水溶液同样处理上述炭,沸腾回流处理2h,然后用纯水洗涤处理后的活性炭至洗涤液的ph值为7~8。烘干得到改性活性炭载体。
41.称取催化剂质量5%的钯2.50g,用王水溶解。用盐酸调节活性钯浸渍液的ph值为0.5,得到活性钯浸渍液。
42.上述改性活性炭载体用去离子水打浆,搅拌条件下稳定1h,得到活性炭浆液。去离
子水用量为每克改性活性炭载体用10ml去离子水。将调节ph值后的活性组分活性钯浸渍液加入活性炭浆液中,搅拌3h,得到催化剂前体。
43.上述催化剂前体陈化、还原、洗涤、干燥条件同实施例1,制得5.0%wt钯炭催化剂。
44.实施例3
45.称取实施例1中相同活性炭50克。放入在烧瓶中。用浓度为10%wt的盐酸溶液,沸腾回流处理2h,然后用纯水洗涤处理后的活性炭至洗涤液的ph值为7~8,烘干。再用1%wt的双氧水溶液同样处理上述炭,沸腾回流处理2h,然后用纯水洗涤处理后的活性炭至洗涤液的ph值为7~8。烘干得到改性活性炭载体。
46.称取催化剂质量5.5%的钯2.75g,用王水溶解。用盐酸调节活性钯浸渍液的ph值为1.0,得到活性钯浸渍液。
47.上述改性活性炭载体用去离子水打浆,搅拌条件下稳定1h,得到活性炭浆液。去离子水用量为每克改性活性炭载体用10ml去离子水。将调节ph值后的活性组分活性钯浸渍液加入活性炭浆液中,搅拌3h,得到催化剂前体。
48.上述催化剂前体陈化、还原、洗涤、干燥条件同实施例1,制得5.5%wt钯炭催化剂。
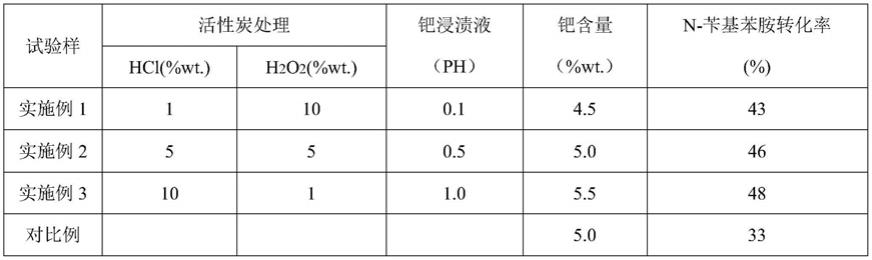
49.对比例
50.市场所购的现有技术生产美罗培南的5%wt钯炭催化剂。
51.催化剂活性试验
52.在培南类药物合成反应中,催化加氢脱苄基类保护基团是钯炭催化剂的一个主要用途。试验采用n-苄基苯胺为原料,对钯炭催化剂进行加氢脱苄试验。该试验与工厂实际情况比较接近,能较好反应钯炭催化剂工业使用的实际性能优劣情况。
53.试验过程:在500ml高压反应釜中,加入0.01g pd对应的钯炭催化剂、30.0gn-苄基苯胺、150ml乙醇、1.0ml甲酸。系统密闭后,先氮气置换空气3次,再用氢气置换氮气3次。排气至常压后开始加热,升温到50℃
±
2℃并保持,通入氢气压力至0.50mpa并保持,调节转速为900r/min,20min后结束试验。关氢,冷却至40℃以下取液体样,气相色谱分析n-苄基苯胺的转化率。
54.催化剂制备工艺条件与加氢脱苄基试验情况见表1。由表1可见采用以本发明技术所制备的钯炭催化剂,脱保护基效率有着显著的提高。
55.表1.催化剂制备工艺与n-苄基苯胺的转化率对照表表1.催化剂制备工艺与n-苄基苯胺的转化率对照表
[0056][0057]
为了说明和描述的目的,已经提供上述对于本发明优选实施方案的描述。但其不意图是穷举的或将本发明限于所公开的确切形式。改变和变化根据上述教导是可能的,或者可通过本发明的实践而获得。选择和描述所述实施方案以解释本发明的原理及其实际应
用,以使本领域技术人员能够以适于预期的具体应用的各种实施方案和各种变型来应用本发明。意图是本发明的范围由所附权利要求书及其等价物限定。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1