复合纳米催化剂的合成方法与流程
1.本发明涉及催化剂合成技术领域,具体公开了一种复合纳米催化剂的合成方法。
背景技术:
2.纳米催化剂因其具有许多优点如较大的表面体积比,更大的活性、选择性和稳定性已经逐渐取代了传统催化剂成为研究的热点,因它们在污染物分离,气体存储、特别是多相催化方面具有巨大潜力,巨表面积的多孔材料越来越收到关注。
3.但是依然有两个主要的问题限制了纳米催化剂的工业化大规模应用,首先,从产物中分离和回收这些细微纳米颗粒依然是不容易的,其次,纳米催化剂非常容易团聚和脱落进而影响了其催化活性。
技术实现要素:
4.本发明的目的在于克服现有技术的缺陷,本发明的目的之一是提供一种复合纳米催化剂的合成方法,用于解决现有技术中类普鲁士蓝纳米催化剂的合成工艺比较繁琐、贵金属负载量低,产生的三废多的问题。
5.为了实现以上目的及其他目的,本发明是通过包括以下技术方案实现的:
6.复合纳米催化剂的合成方法,包括以下步骤:
7.将过渡金属盐溶液与普鲁士蓝类配体溶液在表面活性剂的作用下得到纳米载体;
8.将贵金属盐溶液滴加至所述的纳米载体的溶液中,搅拌均匀后加入还原剂,制得纳米催化剂溶液;以及
9.将所述纳米催化剂溶液去除溶剂,获得所述纳米催化剂。
10.可选地,所述过渡金属盐为乙酸钴、硝酸钴、氯化铁、硝酸铁、氯化铜、氯化镍、氯化锰溶液中的一种。
11.可选地,所述普鲁士蓝类配体溶液为六氰合钴酸钾、赤血盐、黄血盐溶液中的一种。
12.可选地,所述表面活性剂为聚乙烯吡咯烷酮、十六烷基三甲基溴化铵、十六烷基三甲基氯化铵、十二烷基苯磺酸钠、十二烷基硫酸钠、聚乙二醇中的一种。
13.可选地,所述贵金属盐溶液包括氯金酸、氯化钯、四氯铂酸钾溶液中的一种或多种混合物。
14.可选地,所述还原剂为乙醇、甲醇、丙酮、水合肼、硼氢化钠中的一种。
15.可选地,所述纳米催化剂是由所述纳米催化剂溶液水洗后真空干燥所得。
16.可选地,所述纳米载体的溶液干燥时的烤箱温度为55-65℃。
17.可选地,所述纳米催化剂可用于偶联反应和氧化还原反应。
18.可选地,所述纳米催化剂使用后通过离心、过滤后实现分离回收。
19.综上所述,本发明提供了一种复合纳米催化剂的合成方法,该方法不仅具有操作简单,贵金属负载量高,而且采用绿色醇类溶剂还原,三废少,具有很大的经济效益和工业
化应用前景,同时此催化剂可以用于偶连、还原和氧化等反应的催化,且易脱落回收。其他的特征、益处和优势将通过本文详述的包括说明书和权利要求书在内的文本公开而显而易见。
附图说明
20.图1.1是pd-co3[co(cn)6]2的xrd表征图;
[0021]
图1.2是pd-co3[co(cn)6]2的hrtem表征图;
[0022]
图1.3是pd-co3[co(cn)6]2的ftir的表征图;
[0023]
图1.4是pd-co3[co(cn)6]2的xps的能谱图;
[0024]
图1.5是pd-co3[co(cn)6]2的吸附脱曲线图以及粒子孔径分别图;
[0025]
图1.6是pd-co3[co(cn)6]2使用后的转化率、选择率和反应速率图;
[0026]
图1.7是pd-co3[co(cn)6]2使用后sem扫描电镜(扫描电镜)表征途以及xps能谱图;
[0027]
图2.1是pd
x
co
3-x
[co(cn)6]2的xrd表征图;
[0028]
图2.2是pd
x
co
3-x
[co(cn)6]2的hrtem表征图;
[0029]
图2.3是pd
x
co
3-x
[co(cn)6]2的ftir的表征图;
[0030]
图2.4是pd
x
co
3-x
[co(cn)6]2的xps的能谱图;
[0031]
图2.5是pd
x
co
3-x
[co(cn)6]2的吸附脱曲线图以及粒子孔径分别图;
[0032]
图2.6是pd
x
co
3-x
[co(cn)6]2使用后的转化率、选择率和反应速率图。
具体实施方式
[0033]
下面结合具体实施例进一步阐述本发明,应理解,这些实施例仅用于说明本发明而不用于限制本发明的保护范围。
[0034]
以下通过特定的具体实例说明本发明的实施方式,本领域技术人员可由本说明书所揭露的内容轻易地了解本发明的其他优点与功效。本发明还可以通过另外不同的具体实施方式加以实施或应用,本说明书中的各项细节也可以基于不同观点与应用,在没有背离本发明的精神下进行各种修饰或改变。
[0035]
复合纳米催化剂的合成方法,包括以下步骤:
[0036]
将过渡金属盐溶液与普鲁士蓝类配体溶液在表面活性剂的作用下得到纳米载体;
[0037]
将贵金属盐溶液滴加至所述的纳米载体的溶液中,搅拌均匀后加入还原剂,制得纳米催化剂溶液;以及
[0038]
将所述纳米催化剂溶液去除溶剂,获得所述纳米催化剂。
[0039]
实施例1
[0040]
具体地,所述纳米催化剂可以例如为pd-co3[co(cn)6]2催化剂,其合成包括以下步骤:
[0041]
可以选择共沉淀法,即在室温下用注射器将b溶液滴加进a溶液中,10分钟后,在室温下静止24h,然后用蒸馏水将产生的粉红色沉淀过滤和离心清洗3次,放在55-65℃烤箱干燥,例如60℃,收集得到co3[co(cn)6]2固体;
[0042]
其中,所述a溶液是由0.02-0.05mol六氰合钴酸钾,例如0.04mol和0.22-0.35g聚乙烯吡咯烷酮,例如0.3g,在搅拌器中搅拌溶解于10毫升蒸馏水中所得;
[0043]
所述b溶液是由0.06-0.08molco(ch3coo)2·
nh2o(乙酸钴),例如0.075mol,溶解在10毫升蒸馏水中所得;
[0044]
将0.007-0.015克co3[co(cn)6]2,例如0.01克,溶解在20毫升水中,然后将10ml氯化钯溶液(1g/l)滴加入上述混合溶液中,在室温下磁搅拌10分钟后,最后再滴加10ml无水乙醇,四个小时后,离心法收集固体产品用水洗3次并真空干燥得到产物pd-co3[co(cn)6]2。
[0045]
进一步地,采用xrd(x射线衍射)来表征上述所述的复合纳米粒子即pd-co3[co(cn)6]2的物相,如图1.1所示,通过比较负载前后的纳米粒子衍射峰,发现复合纳米粒子多出了2个衍射峰分别对应面心立方pd的111和220晶面,这说明将pd颗粒成功的负载在mof(金属有机物框架)载体上,通过谢乐公式计算,pd纳米晶粒尺寸在3nm左右。
[0046]
进一步地,采用hrtem(高分辨率的透射电镜)表征上述所述的复合纳米粒子即pd-co3[co(cn)6]2,显示pd纳米粒子沉积在co3[co(cn)6]2表面形成核壳结构,如图1.2a所示,很容易观察到co3[co(cn)6]2的直径在100nm左右,如图1.2b所示,通过高分辨率透射电镜图像的单个纳米颗粒可以发现钯纳米粒子封装在微孔隙内,颗粒大小约3nm与xrd结果一致,对该单个纳米粒子进行能量色散x射线分析,如图1.2c所示,发现选中区域包括pd、c、和n元素,说明氯化钯已经成功被还原成pd单质,沉积在mof(金属有机物框架)制备的表面,通过计小颗粒的2晶格条纹如图1.2d所示,得到面间的距离为0.2nm,相对应的pd的(111)晶面,元素地图包括c,n和pd也显示,如图1.2e-h所示,该纳米粒子为核壳结构,pd纳米颗粒负载在co3[co(cn)6]2表面。
[0047]
进一步地,采用ftir(傅氏转换红外线光谱分析仪)表征上述所述的复合纳米粒子即pd-co3[co(cn)6]2,如图1.3所示,2170cm-1
的特征峰在对应于c≡n三键的振动,3645cm-1
和1610cm-1
的特征峰对应其中含有的结晶水振动,相比两个光谱并没有太多的区别,这说明没有明显的基团变化。
[0048]
进一步地,采用xps(x射线荧光探针)能谱描述pd-co3[co(cn)6]2纳米粒子的表面状态,如图1.4所示,可以发现pd和co峰强度几乎差不多,这意味着co3[co(cn)6]2纳米粒子几乎是完全被pd纳米粒子覆盖,这种现象与hrtem中的结论一致,pd 3d的xps谱可以观察到两个主要的双重峰,结合能峰值为335.4ev(pd03d
5/2
)和340.7ev(pd03d
3/2
)可以对应于pd零价态。
[0049]
如图1.1-1.4所示,上述结果表明,pd在co3[co(cn)
6]2
纳米粒子的表面为零价态,另外两个峰对应pd的二价态,是由于一些pd
2+
掺杂到二氧化碳(co(cn)6)2引起的,结合能与k2pd2(cn)4的pd03d
5/2
(338.8ev)和pdⅱ3d
3/2
(334ev)相近,表明pd和c≡n结合;一些pd原子掺杂到co3[co(cn)6]2的晶格中,将纳米粒子由去离子水清洗几次,然后由硝酸溶解做icp(光谱仪)测试,icp(光谱仪)的测试结果显示pd-co摩尔比是2.94:1,而pd-co反应物物质的量比是5:4,足以证明pd
2+
没有吸附而是掺杂。
[0050]
进一步地,当需要检测所述pd-co3[co(cn)6]2纳米粒子孔的性质时,可使用氮气吸附脱附分析研究,在测量之前,样品在60℃真空环境加热10h下以完全脱水,co3[co(cn)6]2的比表面积为459.6m2/g而pd-co3[co(cn)6]2的是470.8m2/g,由于5nm的pd纳米粒子的贡献,使得负载后的比表面积变大,如图1.5a和1.5b所示,两种纳米粒子的吸附脱附曲线很相似,吸附等温线的氮气吸收在低相对压力(p/p0《0.001)下显得很陡峭反映出纳米粒子丰富的微孔结构,一个轻微的回线和等温线在中、高压地区(p/p0=0.8-1.0)急剧上升反应了mof
(金属有机物框架)尺寸的大小,如图1.5c和图1.5d所示,我们可以发现co3[co(cn)6]2在3nm存在,这证明mof(金属有机物框架)的孔隙在3nm左右,此外,另一个孔分布约0.5nm,这符合孔隙大小mii3[miii(cn)6]2多孔框架结构,实际上,大量的微孔利用催化过程中的吸附脱附,大孔则可以加速反应物和产物的传输,合适大小的微孔有利于固定固金属催化剂,pd-co3[co(cn)6]2在3nm处的峰消失了,这表明mof(金属有机框架材料)表面的微孔大多数被pd纳米粒子完全覆盖了。
[0051]
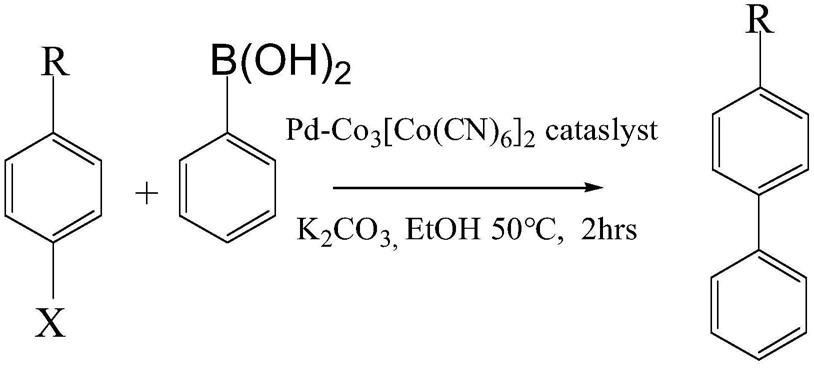
进一步地,当需要测量所述pd-co3[co(cn)6]2催化剂的催化能力时,可以选取suzuki反应(铃木反应)为特征反应,将0.5-2mg pd-co3[co(cn)6]2、0.3-1.3mmol芳香烃卤代物、0.8-1.5mmol苯硼酸和1.5-4mmol碳酸钾加入一个装有30ml乙醇的三口烧瓶中,搅拌回流加热至50℃保持2h后,待反应结束后加入0.5-1.1mg的萘搅拌均匀后,取部分溶液用于气相色谱测量;
[0052]
当测量所述pd-co3[co(cn)6]2催化剂的催化能力时具体地,可以选定1mg pd-co3[co(cn)6]2、1mmol芳香烃卤代物、1.2mmol苯硼酸和3mmol碳酸钾加入一个装有30ml乙醇的三口烧瓶中,搅拌回流加热至50℃保持2h后,待反应结束后加入1mg的萘搅拌均匀得到待检测溶液。
[0053]
如表1所示,即使在室温下,如表1中的1-3所示,碘苯和溴苯都表现出很高的产率,如表1中的4所示,对溴苯甲苯是用来代替溴苯以确认联苯的产生是交叉偶联,而不是苯硼酸自身偶联,结果表明,4-甲基联苯的的产率是99%几乎没有联苯产生;如表1中的5-6所示,氯苯和对氯甲苯产率不是很高,但是通过改变反应条件,如表1中的7-8所示,例如增加反应温度和时间,也可以达到可观的产率,例如,当反应温度80℃,时间为4小时反应物为氯苯,可以获得高达86.2%的产率。这些结果可以达到pd与机膦配体的水平且远高于普通的pd纳米粒子催化剂,同时如表1中的9所示,证明了纯mof(金属有机物框架)没有催化效果,如表1中的10-13所示,同时对于不同取代基的氯苯都有可观的催化能力。
[0054]
表1:苯硼酸与各芳香卤代物的suzuki反应条件与结果
[0055]
[0056][0057]
以pd-co3[co(cn)6]2催化剂为例,pd-co3[co(cn)6]2催化剂的易分离度、重复性和稳定性如下:首先,由于其100nm左右的尺寸,催化剂可以很容易地通过离心分离;其次,如图1.6a所示,8次重复使用后有明显的转化率和选择性降低;如图1.6b所示,前四个循环的动力学研究表明,每个循环的反应速率都没有降低;第三,如表1中的7-8所示,催化剂表现出很好的热稳定性,高的活动在升高的温度下;如图1.7a所示,反应后催化剂的sem(扫描电镜)图像表明,反应后催化剂的相组成和表面形态与之前的结果相比基本没有改变,如图1.7b所示,xps谱高温反应后的催化剂显示没有发生明显的变化,最后,icp-aes(电感耦合等离子体发射光谱)测试结果表明溶液中小于100ppm的pd残留,即pd析出和脱落并没有发生;此外,ice-aes(电感耦合等离子体发射光谱)分析了8个循环后的催化剂的组分没有改变,即pd和co的比率也没有发生变化。
[0058]
实施例2
[0059]
具体地,所述纳米催化剂可以例如为pdxco
3-x
[co(cn)6]2催化剂,其合成包括以下步骤:
[0060]
按照合成pd-co3[co(cn)6]2催化剂步骤中制备co3[co(cn)6]2固体的方法制备co3[co(cn)6]2固体;
[0061]
将0.02-0.07克co3[co(cn)6]2,例如为0.05克,溶解在20毫升水中,然后将10毫升氯化钯溶液(1g/l)滴加入上述混合溶液中并在室温下磁搅拌12h后,收集的固体产品,用水洗3次并真空干燥得到产物pd
x
co
3-x
[co(cn)6]2,例如固体产品的收集方法可以为离心法。
[0062]
进一步地,采用xrd(x射线衍射)来表征上述所述的复合纳米粒子的物相,如图2.1所示,通过比较掺杂前后的纳米粒子衍射峰,没有新的峰产生,这说明将pd被掺杂在mof(金属有机物框架)内。
[0063]
进一步地,如图2.2a所示,采用hrtem(高分辨率的透射电镜)表征上述所述的复合
纳米粒子催化剂,如图2.2b-e所示,元素地图包括co,n和pd,可以发现pd元素信号低于co,n,证明了pd是微量掺杂进入mof(金属有机物框架)中的。
[0064]
进一步地,采用ftir(傅氏转换红外线光谱分析仪)表征上述所述的复合纳米粒子催化剂,如图2.3所示,2170cm-1
的特征峰在对应于c≡n三键的振动,3645cm-1
和1610cm-1
的特征峰对应其中含有的结晶水振动,相比两个光谱并没有太多的区别,这说明没有明显的基团变化。
[0065]
进一步地,采用xps(x射线荧光探针)能谱描述所述的pd
x
co
3-x
[co(cn)6]2纳米粒子的表面状态,如图2.4所示,pd 3d的xps谱可以观察到两个主要的双重峰,结合能峰值为338.8ev(pdⅱ3d
5/2
)和344.1ev(pdⅱ3d
3/2
)可以归因于pd二价态,上述结果表明,pd掺杂入co3[co(cn)6]2纳米粒子的表面为二价态,一些pd原子掺杂到co3[co(cn)6]2的晶格中,将纳米粒子由去离子水清洗几次,然后由硝酸溶解做icp测试,icp的测试结果显示pd-co摩尔比是1:6,而pd-co反应物物质的量比是1:7,足以证明pd
2+
没有吸附而是掺杂。
[0066]
进一步地,可使用氮气吸附脱附分析研究所述的pdxco
3-x[co(cn)6]2纳米粒子的孔性质,具体操作如下:
[0067]
在测量之前,样品在60℃真空环境加热10h使其完全脱水,co3[co(cn)6]2的比表面积为459.6m2/g而pd-co3[co(cn)6]2的是682.3m2/g,如图2.5a和2.5b所示,两种纳米粒子的吸附脱附曲线很相似,吸附等温线的氮气吸收在低相对压力(p/p0《0.001)下显得很陡峭反映出纳米粒子丰富的微孔结构,一个轻微的回线和等温线在中、高压地区(p/p0=0.8-1.0)急剧上升,反应了mof(金属有机物框架)尺寸的大小;如图2.5c和2.5d所示,可以发现二者在3nm存在微孔,这证明mof(金属有机物框架)的孔隙在3nm左右,此外,另一个孔分布约0.5nm,这符合的孔隙大小mii3[miii(cn)6]2多孔框架结构,大量的微孔利用催化过程中的吸附脱附,大孔则可以加速反应物和产物的传输,合适大小的微孔有利于固定金属催化剂。
[0068]
进一步地,当需要测量所述pd-co3[co(cn)6]2催化剂的催化能力时,可以选取suzuki反应(铃木反应)为特征反应,将0.05-0.15mol%pdxco
3-x[co(cn)6]2、0.3-1.3mmol芳香烃卤代物、0.8-1.5mmol苯硼酸和1.5-4mmol碳酸钾加入一个装有30ml乙醇的三口烧瓶中,搅拌回流加热至50℃保持2h后,待反应结束后加入0.5-1.1mg的萘搅拌均匀后,取部分溶液用于气相色谱测量;
[0069]
具体地,可以选取0.1mol%pd
x
co
3-x
[co(cn)6]2、1mmol芳香烃卤代物、1.2mmol苯硼酸和3mmol碳酸钾加入一个装有30ml乙醇的三口烧瓶中,搅拌回流加热至50℃保持2h后,待反应结束后加入1mg的萘搅拌均匀得到待检测溶液。
[0070]
如表2中的1-4所示,即使在室温下,碘苯和溴苯都表现出很高的产率,如表2中的5所示,对溴苯甲苯是用来代替溴苯以确认联苯的产生是交叉偶联,而不是苯硼酸自身偶联,结果表明,4-甲基联苯的的产率是99%几乎没有联苯产生,如表2中的6-7所示,氯苯和对氯甲苯的产率不是很高,但是通过改变反应条件,如增加反应温度和时间,如表2中的8-9所示,也可以达到可观的产率,这些结果可以达到pd与机膦配体的水平且远高于普通的pd纳米粒子催化剂,如表2中的10所示,我们证明了纯mof(金属有机物框架)没有催化效果。
[0071]
表2.:苯硼酸与各芳香卤代物的suzuki反应条件与结果
[0072][0073][0074]
pd
x
co
3-x
[co(cn)6]2催化剂的易分离度、重复性和稳定性如下:首先,由于其100nm左右的尺寸,催化剂可以很容易地通过离心分离,其次,如图2.6a所示,8次重复使用后有明显的转化率和选择性降低;此外,如图2.6b所示,前四个循环的动力学研究表明,每个循环的反应速率都没有降低。
[0075]
实施例3
[0076]
具体地,所述纳米催化剂可以例如为pt-fe3[fe(cn)6]2催化剂,其合成包括以下步骤:
[0077]
在室温下用注射器将b溶液滴加进a溶液中,10分钟后,在室温下静止24h,然后用蒸馏水将产生的粉红色沉淀过滤和离心清洗3次,放在55-65℃烤箱干燥,例如为60℃,收集得到fe3[fe(cn)6]2固体;
[0078]
其中,所述a溶液是由0.03-0.09mol赤血盐,例如0.06mol和0.1-0.4g聚乙烯吡咯烷酮,例如0.3g,在搅拌器中搅拌溶解于10毫升蒸馏水中所得;
[0079]
所述b溶液是由0.02-0.07mol,例如0.05mol氯化铁溶解在10毫升蒸馏水中所得;
[0080]
将3-8克,例如5克上述所得fe3[fe(cn)6]2溶解在200毫升水中,然后将100毫升四氯铂酸钾溶液(1g/l)滴加入上述混合溶液在室温下磁搅拌10分钟后,最后再滴加10ml无水乙醇,四个小时后收集的固体产品,用水洗3次并真空干燥得到产物pt-fe3[fe(cn)6]2,例如可以用离心法收集。
[0081]
进一步地,使用所述的pt-fe3[fe(cn)6]2纳米催化剂制备2,4-二氯-5-异丙氧基苯胺胺醚的方法包括如下步骤:
[0082]
在容量为1~2l的反应釜中加入2-3g所述的pt-fe3[fe(cn)6]2纳米催化剂,例如为
2.6g,并加入48~53ml的氨水,例如为51ml,并开启搅拌,使两者充分混合均匀;
[0083]
搅拌20-30分钟后,向反应釜内加入392~400ml的乙醇,例如为396ml,和247~251g的2,4-二氯-5-异丙氧基硝基苯、例如为249g;
[0084]
向反应釜内通入氮气,将反应釜内的空气置换掉,然后再过8~10分钟后,关闭氮气的通入,再向反应釜内通入氢气,用以置换反应釜内的氮气,再过8~10分钟后关闭氢气的通入,再次向反应釜内通入氮气来置换氢气,反复三次后,保持反应釜内氢气压力为0.5~1mpa,升温至80~85℃,反应6~8小时;
[0085]
反应结束后,采用离心分离的方法,将催化剂分离,并将反应釜减压,蒸馏出溶剂,得到灰色固体212~216g,即2,4-二氯-5-异丙氧基苯胺;可选的,反应釜内氢气压力为0.7mpa,升温至82℃,反应7.2小时,得到214.5g2,4-二氯-5-异丙氧基苯胺。
[0086]
通过上述方法获得的2,4-二氯-5-异丙氧基苯胺,含量为99%,收率为98.8%。
[0087]
综上所述,采用本发明的合成方法获得纳米粒子催化剂不仅合成方法简单、对偶连反应、氧化反应具有极高的催化活性,而且还具有易分散、易分离、易回收、高稳定性等特点,此纳米催化剂具有比表面积大、暴露表面的配位不饱和的活性位点多的特点,因而在催化反应过程中,具有明显优异的催化活性,而纳米催化剂在回收过程中,可以在离心和过滤实现简单分离,回收套用,快捷方便,而且回收套用次数更是达到10次及以上,节约了成本。因此,本发明具有高度的产业利用价值。
[0088]
上述实施例仅例示性说明本发明的原理及其功效,而非用于限制本发明。任何熟悉此技术的人士皆可在不违背本发明的精神及范畴下,对上述实施例进行修饰或改变。因此,举凡所属技术领域中具有通常知识者在未脱离本发明所揭示的精神与技术思想下所完成的一切等效修饰或改变,仍应由本发明的权利要求所涵盖。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1