一种石墨相氮化碳负载低自旋单原子Fe的多相催化剂、制备方法及催化方法
一种石墨相氮化碳负载低自旋单原子fe的多相催化剂、制备方法及催化方法
技术领域
1.本发明涉及乙醇催化转化及相关催化技术领域,尤其涉及一种石墨相氮化碳负载低自旋单原子fe的多相催化剂、制备方法及催化方法。
背景技术:
2.噻菌灵又称噻苯咪唑,是一种重要的苯咪唑类广谱杀菌剂,它的合成可以采用2-(1-羟基乙基)-苯并咪唑为原料按照如下路线1中的反应步骤合成(化学试剂,2016,38,1235-1238)。
[0003][0004]
其中,2-(1-羟基乙基)-苯并咪唑作为噻菌灵的重要前体,其工业及实验室制备方法如路线2所示,由邻苯二胺和乳酸为反应物,高浓度盐酸为催化剂,回流的条件下经缩合反应先制得2-(1-羟基乙基)-苯并咪唑盐酸盐后,用氨水去质子化得2-(1-羟基乙基)-苯并咪唑(org. chem.front.,2019,6,205-208)。
[0005][0006]
但此路线的缺陷是用浓盐酸作为催化剂,具有腐蚀性,而且该反应在高温下进行,回流温度在80℃以上,容易发生安全隐患。
[0007]
因此,寻求一种反应过程温和、环境友好的用于制备2-(1-羟基乙基)-苯并咪唑的催化剂及催化反应路线,是亟需解决的反应。
技术实现要素:
[0008]
为了解决上述问题,本发明提供一种石墨相氮化碳负载低自旋单原子fe的多相催化剂,并首次将其用于催化乙醇和苯并咪唑反应制备2-(1-羟基乙基)-苯并咪唑。所述多相催化剂由均匀分散的单原子fe与石墨相氮化碳载体组成,通过控制制备过程条件,得到占比不同的低自旋状态,从而影响催化反应收率。
[0009]
本发明的技术方案如下:
[0010]
本发明提供一种石墨相氮化碳负载低自旋单原子fe的多相催化剂,所述多相催化剂由均匀分散的单原子fe与石墨相氮化碳载体组成,其中,
[0011]
所述石墨相氮化碳载体的碳氮元素比为c/n=0.4~0.7,所述fe与所述石墨相氮化碳载体表面的四个n原子形成fe-n四配位结构,fe-n的键长为,单原子fe的负载量为3wt%~10wt%。
[0012]
进一步地,所述多相催化剂中,低自旋状态的单原子fe占单原子fe总数的20~90%。
[0013]
进一步地,所述多相催化剂中,低自旋状态的单原子fe占单原子fe总数的80%。
[0014]
本发明还提供一种前述多相催化剂的制备方法,在本发明的一个具体的实施方式中,所述方法包括如下步骤:
[0015]
步骤a1:分别配制双氰胺溶液和fe(iii)盐溶液,所述双氰胺溶液的浓度为0.4~0.8mol/l,所述fe(iii)盐溶液的浓度为0.03~0.06mol/l,在常温下缓慢地将铁盐溶液倒入双氰胺溶液中,得到深红色的fe(iii)-双氰胺配合物溶液,将所述配合物溶液倒入蒸发皿中,将水蒸干,得到土黄色的fe(iii)-双氰胺配合物片状固体;
[0016]
步骤b1:将所述fe(iii)-双氰胺配合物片状固体研磨成细粉后置于管式炉中,以 5℃~15℃/min的升温速率升温至450~650℃,惰性气体气氛下热处理2~3h后自然降温,所得产物研磨均匀后分散于稀hno3溶液中,在磁力搅拌下,将所得悬浮液在40℃~60℃温度下保持4~6h,过滤,洗涤滤液呈中性,在40℃~60℃温度的真空干燥箱中真空干燥4~8h,得 fe-n-gcn-sas-ls-原位碳化法,意为原位碳化法制备石墨相氮化碳负载单原子fe。
[0017]
其中,gcn:石墨相氮化碳,sas:单原子,ls:低自旋状态。
[0018]
进一步地,步骤a1中,所述fe(iii)盐为fe(no3)3·
9h2o。
[0019]
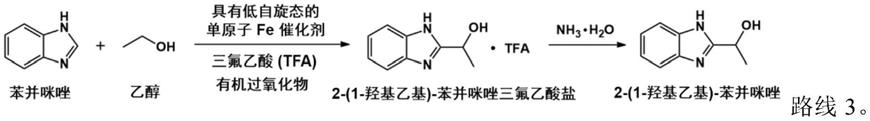
进一步地,步骤a1中,于90℃温度下将水蒸干。
[0020]
进一步地,步骤b1中,所述惰性气氛可选自热处理气氛选自n2/he/ar。
[0021]
进一步地,步骤b1中,所述稀hno3溶液的浓度为1.5~2.5mol/l。
[0022]
进一步地,步骤b1中,所述磁力搅拌的频率为300~500rpm。
[0023]
进一步地,步骤b1中,所述洗涤用去离子水和95%乙醇洗涤进行。
[0024]
进一步地,步骤b1中,所述管式炉升温至550~600℃。
[0025]
所述石墨相氮化碳的温度在450~650℃是稳定的,优选地,在550~600℃下是最稳定的。在此温度下,fe(iii)双氰胺配合物原位碳化成石墨相氮化碳,温度的不同影响了fe和石墨相氮化碳的结合方式以及低自旋态。在550~600℃发生原位碳化时,低自旋单原子fe占单原子 fe原子总数的比例更高。
[0026]
在本发明的另一个具体实施方式中,所述方法包括如下步骤:
[0027]
步骤a2:称取双氰胺(m=84.08g/mol)在管式炉中以5℃~15℃/min的升温速率升温至 450~650℃,n2气氛下热处理2~3h后自然降温可得黄色固体,为石墨相氮化碳gcn;
[0028]
步骤b2:取gcn黄色固体加入圆底旋转蒸发瓶中,将铁盐溶于无水乙醇后倒入所述旋转蒸发瓶中,于40℃~60℃温度下减压旋蒸20min~40min得干燥的黄色固体,将黄色固体后置于管式炉中,以5℃~15℃/min的升温速率升温至450~650℃,惰性气体气氛下热处理2~3h后自然降温,所得产物研磨均匀后分散于稀hno3溶液中,在磁力搅拌下,将所得悬浮液在 40℃~60℃温度下保持4~6h,过滤,洗涤直到滤液呈中性,在40℃~60℃温度下的真空干燥箱中真空干燥4~8h,得fe-n-gcn-sas-ls-浸渍法,意为浸渍法制备石墨相氮化碳负载单原子fe。
[0029]
其中,gcn:石墨相氮化碳,sas:单原子,ls:低自旋状态。
[0030]
进一步地,步骤b2中,所述fe(iii)盐为fecl3·
9h2o。
[0031]
进一步地,步骤b2中,无水乙醇中,fe(iii)的浓度为0.1~0.3mol/l。
[0032]
进一步地,步骤b2中,所述管式炉升温至550~600℃。
[0033]
进一步地,步骤b2中,所述惰性气氛可选自热处理气氛选自n2/he/ar。
[0034]
进一步地,步骤b2中,所述稀hno3溶液的浓度为1.5~2.5mol/l。
[0035]
进一步地,步骤b2中,所述磁力搅拌的频率为300~500rpm。
[0036]
进一步地,步骤b2中,所述洗涤用去离子水和95%乙醇洗涤进行。
[0037]
本发明还提供利用前述多相催化剂用于乙醇和苯并咪唑制备2-(1-羟基乙基)-苯并咪唑的催化方法,经如下反应路线3所示,采用乙醇和苯并咪唑作为反应原料,以有机过氧化物为介导,具有低自旋状态的单原子fe作为催化活性中心,制备2-(1-羟基乙基)-苯并咪唑,其中,具有低自旋状态的单原子fe催化剂为所述石墨相氮化碳负载低自旋单原子fe的多相催化剂。
[0038][0039]
进一步地,所述催化方法包括如下步骤:
[0040]
在反应容器中加入苯并咪唑(m=118.14g/mol),加入无水乙醇使其充分溶解,加入基于苯并咪唑摩尔量计算1.5~2.5倍当量的三氟乙酸,加入含有基于苯并咪唑摩尔量计算的4~10 mol%fe的所述多相催化剂,将体系中的空气抽除后通入n2保护,在n2气体保护下注入以苯并咪唑摩尔量计算4~8倍当量的叔丁基过氧化氢(tbhp),反应于20~40℃下、在磁力搅拌下进行6~18h;
[0041]
反应完毕后过滤分离所述多相催化剂,将所得滤液于40~60℃温度下旋蒸得少量棕色液体,加入碱除酸并调节ph=9~10使反应产物去质子化,提纯得目标产物2-(1-羟基乙基)-苯并咪唑。
[0042]
进一步地,所述反应容器为schlenk瓶,为一种便于抽真空、充惰性气体而设计的带活塞支管的玻璃仪器。
[0043]
进一步地,所述磁力搅拌的频率为300~500rpm。
[0044]
进一步地,所述碱为氨水。
[0045]
进一步地,所述提纯为加入少量甲醇过柱提纯。
[0046]
本发明的有益效果如下:
[0047]
1.本发明的多相催化剂由单原子fe与石墨相氮化碳载体组成,载体碳氮元素含量以及单原子fe的自旋状态可调控,其中制备出具有高达约80%低自旋状态的单原子fe催化剂用于催化乙醇和苯并咪唑反应反应可获得87%的2-(1-羟基乙基)-苯并咪唑收率,首次报道了乙醇和苯并咪唑反应制备2-(1-羟基乙基)-苯并咪唑的反应,是本发明的创新之处。
[0048]
2.本发明采用两种反应路径制备多相催化剂。步骤a1和b1采用了fe和载体原位碳化一步制备方案。步骤a2和b2采用先制备gcn,再将单原子fe负载在gcn载体上的顺序。两种不同的制备路径影响了fe和载体的结合方式,进而影响fe低自旋状态和占比。低自旋状态占比越高,活性中心为越多,催化活性越高。
[0049]
3.所述多相催化剂在反应过程中保持稳定,可循环使用5次及以上。
[0050]
4.所述催化反应条件温和,该反应可在常温(25℃)下进行,相比于目前工业上采
用的盐酸催化邻苯二胺和乳酸缩合法(100℃),反应过程更加安全且环境友好。
附图说明
[0051]
图1本发明实施例1~3中合成的fe-n-gcn-sas-ls系列催化剂的ft-ir图;其中横坐标为波数,单位:cm-1
;纵坐标为透过率。
[0052]
图2本发明实施例1~3中合成的fe-n-gcn-sas-ls系列催化剂的haadf-stem图 (haadf-stem:高角环形暗场-扫描透射电子显微镜)。
[0053]
图3本发明实施例1~3中合成的fe-n-gcn-sas-ls系列催化剂的exafs拟合结果(exafs:扩展x射线吸收精细结构)。
[0054]
图4本发明实施例1中合成的fe-n-gcn-sas-ls-80催化剂的结构示意图。
[0055]
图5本发明实施例1~3中合成的fe-n-gcn-sas-ls系列催化剂的穆斯堡尔谱图;其中横坐标为多普勒速度,单位:cm-1
;纵坐标为γ射线计数率。
[0056]
图6为2-(1-羟基乙基)-苯并咪唑的核磁共振氢谱1h-nmr(左)和核磁共振碳谱
13
c-nmr(右)。
具体实施方式
[0057]
下面结合实施例对本发明做进一步说明,但本发明并不限于以下实施例。
[0058]
实施例1
[0059]
一种石墨相氮化碳负载低自旋单原子fe的多相催化剂,所述多相催化剂由均匀分散的单原子fe与石墨相氮化碳载体组成,其中,
[0060]
所述石墨相氮化碳载体的碳氮元素比为c/n=0.5,所述fe与所述石墨相氮化碳载体表面的四个n原子形成fe-n四配位结构,fe-n的键长为,单原子fe的负载量为8.0wt%,低自旋状态的单原子fe占单原子fe总数的80%。
[0061]
制备过程:
[0062]
步骤a1:称取24.0g双氰胺(m=84.08g/mol)溶于400ml去离子水,称取9.0gfe(no3)3·
9h2o (m=404.0g/mol)溶于100ml去离子水,25℃下缓慢地将硝酸铁溶液倒入双氰胺溶液,得深红色的fe(iii)-双氰胺配合物溶液,将配合物溶液倒入蒸发皿中,于70℃下将水蒸干,得土黄色的fe-双氰胺配合物片状固体;
[0063]
步骤b1:将fe(iii)-双氰胺配合物片状固体研磨成细粉后置于管式炉中,以10℃/min的升温速率升温至580℃,n2气氛下热处理2h后自然降温,所得产物研磨均匀后分散于2mol/l的稀hno3溶液中,在400rpm磁力搅拌下,将所得悬浮液在50℃下保持6h,过滤,用去离子水和95%乙醇洗涤直到滤液呈中性,在40℃的真空干燥箱中真空干燥12h,得 fe-n-gcn-sas-ls-80-原位碳化法,意为原位碳化法制备石墨相氮化碳负载单原子fe,低自旋状态的单原子fe占单原子fe总数约80%。
[0064]
催化方法:
[0065]
用实施例1所得到的多相催化剂催化苯并咪唑与乙醇制备2-(1-羟基乙基)-苯并咪唑。在 schlenk瓶中加入1.0mmol的苯并咪唑(m=118.14g/mol),加入4ml的无水乙醇使其充分溶解,加入以苯并咪唑为基准2倍当量的三氟乙酸,35mgfe-n-gcn-sas-ls-80催化剂(基于苯并咪唑摩尔量计算,共含5mol%fe),将体系中的空气抽除后通入n2保护,在n2气体
保护下注入以苯并咪唑为基准5倍当量的叔丁基过氧化氢(tbhp),反应于25℃、400rpm磁力搅拌下进行12h;
[0066]
反应完毕后过滤分离多相催化剂,将所得滤液于40℃下旋蒸得少量棕色液体,加入氨水除酸并调节ph=9~10使反应产物去质子化,加入少量甲醇后过柱提纯得目标产物2-(1-羟基乙基)-苯并咪唑,收率87%。
[0067]
实施例2
[0068]
一种石墨相氮化碳负载低自旋单原子fe的多相催化剂,所述多相催化剂由均匀分散的单原子fe与石墨相氮化碳载体组成,其中,
[0069]
所述石墨相氮化碳载体的碳氮元素比为c/n=0.6,所述fe与所述石墨相氮化碳载体表面的四个n原子形成fe-n四配位结构,fe-n的键长为,单原子fe的负载量为4.0wt%,低自旋状态的单原子fe占单原子fe总数的40%。
[0070]
制备过程:
[0071]
步骤a1:同实施例1;
[0072]
步骤b1:将fe(iii)-双氰胺配合物片状固体研磨成细粉后置于管式炉中,以10℃/min的升温速率升温至480℃,n2气氛下热处理2h后自然降温,所得产物研磨均匀后分散于2mol/l的稀hno3溶液中,在400rpm磁力搅拌下,将所得悬浮液在50℃下保持6h,过滤,用去离子水和95%乙醇洗涤直到滤液呈中性,在40℃的真空干燥箱中真空干燥12h,得 fe-n-gcn-sas-ls-40-原位碳化法,意为原位碳化法制备石墨相氮化碳负载单原子fe,低自旋状态的单原子fe占单原子fe总数约40%。
[0073]
催化方法:
[0074]
用实施例2所得到的多相催化剂催化苯并咪唑与乙醇制备2-(1-羟基乙基)-苯并咪唑。在 schlenk瓶中加入1.0mmol的苯并咪唑(m=118.14g/mol),加入4ml的无水乙醇使其充分溶解,加入以苯并咪唑为基准2倍当量的三氟乙酸,70mgfe-n-gcn-sas-ls-40催化剂(含有基于苯并咪唑摩尔量计算的5mol%fe),将体系中的空气抽除后通入n2保护,在n2气体保护下注入以苯并咪唑为基准5倍当量的叔丁基过氧化氢(tbhp),反应于25℃、400rpm磁力搅拌下进行12h。反应完毕后过滤分离多相催化剂,将所得滤液于40℃下旋蒸得少量棕色液体,加入氨水除酸并调节ph=9~10使反应产物去质子化,加入少量甲醇后过柱提纯得目标产物2-(1-羟基乙基)-苯并咪唑,收率67%。
[0075]
实施例3
[0076]
一种石墨相氮化碳负载低自旋单原子fe的多相催化剂,所述多相催化剂由均匀分散的单原子fe与石墨相氮化碳载体组成,其中,
[0077]
所述石墨相氮化碳载体的碳氮元素比为c/n=0.6,所述fe与所述石墨相氮化碳载体表面的四个n原子形成fe-n四配位结构,fe-n的键长为,单原子fe的负载量为6.0wt%,低自旋状态的单原子fe占单原子fe总数的25%。
[0078]
制备过程:
[0079]
步骤a2:称取24.0g双氰胺(m=84.08g/mol)在管式炉中以10℃/min的升温速率升温至550℃,n2气氛下热处理2h后自然降温可得黄色固体,为石墨相氮化碳gcn;
[0080]
步骤b2:取2.0g黄色固体加入圆底旋转蒸发瓶,称取0.96gfecl3·
6h2o(m=270.3g/mol)溶于20ml无水乙醇后倒入步骤a中所述旋转蒸发瓶,于40℃下减压旋蒸30min
得干燥的黄色固体,将黄色固体后置于管式炉中,以10℃/min的升温速率升温至550℃,n2气氛下热处理2h后自然降温,所得产物研磨均匀后分散于2mol/l的稀hno3溶液中,在400rpm磁力搅拌下,将所得悬浮液在50℃下保持6h,过滤,用去离子水和95%乙醇洗涤直到滤液呈中性,在40℃的真空干燥箱中真空干燥12h,得fe-n-gcn-sas-ls-25-浸渍法,意为浸渍法制备石墨相氮化碳负载单原子fe,低自旋状态的单原子fe占单原子fe总数约25%。
[0081]
催化方法:
[0082]
用实施例3所得到的多相催化剂催化苯并咪唑与乙醇制备2-(1-羟基乙基)-苯并咪唑。在 schlenk瓶中加入1.0mmol的苯并咪唑(m=118.14g/mol),加入4ml的无水乙醇使其充分溶解,加入以苯并咪唑为基准2倍当量的三氟乙酸,47mgfe-n-gcn-sas-ls-25催化剂(基于苯并咪唑摩尔量计算的5mol%fe),将体系中的空气抽除后通入n2保护,在n2气体保护下注入以苯并咪唑为基准5倍当量的叔丁基过氧化氢(tbhp),反应于25℃、400rpm磁力搅拌下进行12h。反应完毕后过滤分离催化剂,将所得滤液于40℃下旋蒸得少量棕色液体,加入氨水除酸并调节ph=9~10使反应产物去质子化,加入少量甲醇后过柱提纯得目标产物2-(1
‑ꢀ
羟基乙基)-苯并咪唑,收率38%。
[0083]
如图1所示,图1为本发明实施例1~3合成的石墨相氮化碳负载单原子fe催化剂与未负载fe样品的ft-ir图,图中1556cm-1
对应于石墨相氮化碳三嗪结构碳氮双键的伸缩振动, 893cm-1
与810cm-1
谱带对应于石墨相氮化碳的三嗪结构的平面外弯曲振动,该表征证明所合成的载体结构为石墨相氮化碳。
[0084]
如图2所示,图2为本发明实施例1~3合成的石墨相氮化碳负载单原子fe催化剂的 haadf-stem图,图中的亮点代表复合氧化物负载原子级分散的fe,很明显可以看出,实施例1~3所制备催化剂中的fe为单原子均匀分散的状态。
[0085]
如图3所示,图3为本发明实施例1~3合成的石墨相氮化碳负载单原子fe催化剂的 exafs拟合结果,该结果由exafs的原始数据经拟合计算得到,由拟合结果可知单原子fe 的形成是由于fe与载体形成了fe-n配位。以fe-n-gcn-sas-ls-80催化剂为例,其fe-n 键的键长为,fe与载体表面的四个氮原子形成了fe-n四配位结构,该结构的示意图如图4所示,黑色的为铁原子,黄色的为碳原子,蓝色的为氮原子。
[0086]
如图5所示,图5为本发明实施案例1~3合成的fe-n-gcn-sas-ls系列催化剂的穆斯堡尔谱图,其中箭头所指的位置为具有低自旋状态的fe中心,该具有低自旋状态的fe中心在三个样品中的所有fe物种中,占比分别约为80%,40%及25%,可由穆斯堡尔谱拟合计算得到。由晶体场理论可知,低自旋状态fe中心的形成是由于形成了内轨型fe-n四配位结构,填满了t
2g
(d
xy
,d
xz
,d
yz
)低能三重简并态的电子轨道并空出了eg(d
x2-y2
,d
z2
)高能双重简并态的电子轨道。eg轨道由于能量较高,易于接受孤对电子,因此同时促进了叔丁基过氧化氢和乙醇分子的活化,由反应条件可知促进了乙醇与苯并咪唑制备2-(1-羟基乙基)-苯并咪唑的反应。
[0087]
如图6所示,所得2-(1-羟基乙基)-苯并咪唑为白色固体,其核磁数据为:
[0088]1h nmr(600mhz,dmso-d6):δ12.24(s,1h),7.49(d,j=8.4hz,2h),7.13(dd,j=5.4,2.4hz, 2h),5.78(d,j=4.8hz,1h),4.97-4.93(m,1h),1.52(d,j=6.6hz,3h).
[0089]
13
c nmr(150mhz,dmso-d6):δ158.53,143.04,134.12,121.44,120.92,118.37,111.21,63.66, 22.95.
[0090]
实施例1-3中的性能数据证明,实施例1中所述石墨相氮化碳负载80%低自旋状态的单原子fe催化剂fe-n-gcn-sas-ls-80为最优选择。
[0091]
对比例1
[0092]
用四苯基卟啉氯化铁(m=704.02g/mol)催化苯并咪唑与乙醇制备2-(1-羟基乙基)-苯并咪唑。在schlenk瓶中加入1.0mmol的苯并咪唑(m=118.14g/mol),加入4ml的无水乙醇使其充分溶解,加入以苯并咪唑为基准2倍当量的三氟乙酸,35mg四苯基卟啉fe(共含5mol% fe),将体系中的空气抽除后通入n2保护,在n2气体保护下注入以苯并咪唑为基准5倍当量的叔丁基过氧化氢(tbhp),反应于25℃、摇床振荡下进行12h。反应完毕后旋蒸得少量红色液体,加入氨水除酸并调节ph=9~10使反应产物去质子化,加入少量甲醇后过柱提纯得目标产物2-(1-羟基乙基)-苯并咪唑,收率54%。
[0093]
由实施例和对比例1的反应结果可知,实施例1中所述的多相单原子fe催化剂 fe-n-gcn-sas-ls-80相比于均相配合物四苯基卟啉氯化铁具有更好的催化苯并咪唑与乙醇制备2-(1-羟基乙基)-苯并咪唑的性能。
[0094]
对比例2
[0095]
用四苯基卟啉铜(m=676.27g/mol)催化苯并咪唑与乙醇制备2-(1-羟基乙基)-苯并咪唑。在schlenk瓶中加入1.0mmol的苯并咪唑(m=118.14g/mol),加入4ml的无水乙醇使其充分溶解,加入以苯并咪唑为基准2倍当量的三氟乙酸,35mg四苯基卟啉fe(共含5mol%fe),将体系中的空气抽除后通入n2保护,在n2气体保护下注入以苯并咪唑为基准5倍当量的叔丁基过氧化氢(tbhp),反应于25℃、摇床振荡下进行12h。反应完毕后旋蒸得少量红色液体,加入氨水除酸并调节ph=9~10使反应产物去质子化,加入少量甲醇后过柱提纯得目标产物2-(1-羟基乙基)-苯并咪唑,收率5%。
[0096]
由实施例、对比例1和对比例2的反应结果可知,fe催化剂在催化苯并咪唑与乙醇制备 2-(1-羟基乙基)-苯并咪唑的性能远强于cu催化剂。
[0097]
以上所述,仅是本发明的较佳实施例而已,并非是对本发明作任何其他形式的限制,而依据本发明的技术实质所作的任何修改或等同变化,仍属于本发明所要求保护的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1