一种超分子手性纳米催化剂及其制备方法和应用
1.本发明手性催化剂技术领域,具体涉及一种超分子手性纳米催化剂及其制备方法和应用。
背景技术:
2.长期以来,人们主要靠从自然界获取手性化合物。随着现代社会对手性化合物需求的急剧增加,天然手性化合物无论从种类还是数量上都已远远不能满足这一需求,利用化学方法获取手性化合物成为必然的选择。但人们在进行化学合成时,往往得不到单一的单手性物质,而是得到一对对映体的等量混和物。手性催化合成是获得手性物质的最有效的方法。手性催化合成的核心是手性催化剂。手性催化剂一般为金属配合物手性催化剂,即手性配体与金属离子络合形成的金属有机化合物。金属有机配合手性催化目前取得了长足的发展,但存在着很多问题需要解决。例如手性配体的合成较为复杂,手性原料种类稀少且价格昂贵,并且多数手性催化剂转化数较低,稳定性不高,难以回收和重复使用等。因此,开发和发展新型的手性催化剂,尤其是不含手性小分子的超分子手性催化剂,具有重要的理论意义和实际应用前景。
技术实现要素:
3.针对现有技术中的不足之处,本发明基于导电高分子超分子手性聚苯胺(pani)和cu
2+
催化活性的功能复合,构建出了一种具有优异手性选择性的超分子手性纳米催化剂,其可用于选择性催化一种构型的多巴分子。
4.为实现上述目的,本发明提供如下技术方案:
5.一种超分子手性纳米催化剂的制备方法,包括如下步骤:
6.s1:以单手性小分子樟脑磺酸为掺杂剂和诱导剂合成单手性聚苯胺,以 r型樟脑磺酸诱导苯胺低聚物和苯胺单体合成r-pani-csa纳米纤维,以s 型樟脑磺酸诱导苯胺低聚物和苯胺单体合成s-pani-csa纳米纤维;
7.s2:分别通过氨水溶液对单手性聚苯胺进行去掺杂,得到具有超分子手性的r-pani-undoped和s-pani-undoped纳米纤维;
8.s3:通过非手性分子巯基乙酸对步骤s2产物进行再掺杂得到超分子手性 r-pani-ta和s-pani-ta纳米纤维;
9.s4:将s3的产物在氯化铜溶液中吸附铜离子形成超分子手性 r-pani-ta@cu
2+
和s-pani-ta@cu
2+
纳米纤维。
10.优选地,所述s1的具体制备过程为:配制溶液a:樟脑磺酸、苯胺低聚物和苯胺单体按照质量比为150~301:1:9~17溶解在超纯水中,超声溶解均匀;配制溶液b:0.4~0.5g/ml过硫酸铵水溶液;在溶液a搅拌情况下加入溶液b,搅拌30s后静置,然后每间隔0.5h加样一次,最后静置24h后超纯水离心洗样3次,即得,过硫酸铵与苯胺低聚物的质量比为21~42:1。
11.优选地,所述氨水的浓度为0.1mol/l。
12.优选地,所述巯基乙酸使用时配制成水溶液,浓度为1m。
13.优选地,所述氯化铜水溶液的浓度为1
×
10^-4
m。
14.本案提供一种如上所述的制备方法制得的超分子手性纳米催化剂。
15.进一步地,本案提供一种如上所述的超分子手性纳米催化剂的应用,用于选择性催化一种构型的多巴分子。
16.本发明的有益效果是:本发明提供的超分子手性pani@cu
2+
手性催化剂可以选择性的催化一种构型的dopa分子,超分子手性r-pani(ta)cu
2+
催化剂对r-dopa反应更快;而超分子手性s-pani(ta)cu
2+
催化剂对s-dopa反应更快。同时超分子手性催化剂不含有手性小分子,仅仅依靠聚苯胺的超分子手性组装即可实现良好的不对称催化效果,极大拓宽了不对称催化剂的设计思路,同时也有助于人们更加深入的理解手性来源这个重大科学问题。并且,本案所设计的手性催化剂稳定性高,通过简单离心操作即可进行回收及重复利用。
附图说明
17.为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
18.图1为实施例1-3各产物的tem图(a1-a3分别对应s-pani(csa)、 r-pani(csa)和pani(hcl);b1-b3分别对应s-pani(undoped)、 r-pani(undoped)和pani(undoped);c1-c3分别对应s-pani(ta)、r-pani(ta)和pani(ta))。
19.图2为实施例1-3中各产物的uv光谱图。
20.图3为实施例1-3中各产物的cd光谱图。
21.图4为s-pani(ta)@cu
2+
、r-pani(ta)@cu
2+
和pani(ta)@cu
2+
的sem 图。
22.图5为对应图4的局部放大图以及理论模型图。
23.图6为s-pani(ta)@cu
2+
、r-pani(ta)@cu
2+
和pani(ta)@cu
2+
的tem 图。
24.图7为实施例1-3制得的超分子手性催化剂的紫外光谱图和cd光谱图((a)为s-pani(ta)@cu
2+
、r-pani(ta)@cu
2+
和pani(ta)@cu
2+
的紫外光谱图;(b)为s-pani(ta)@cu
2+
、r-pani(ta)@cu
2+
和pani(ta)@cu
2+
的cd 谱图)。
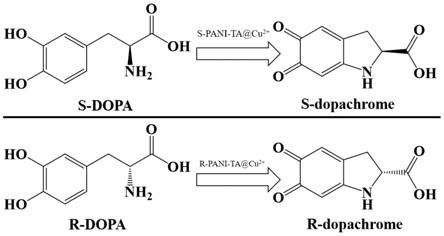
25.图8分别为s-dopa和r-dopa在催化剂作用下转化成手性产物的分子式示意图。
26.图9(a-c)分别为s-pani(ta)@cu
2+
、r-pani(ta)@cu
2+
和 pani(ta)@cu
2+
对s-dopa或r-dopa为底物的动力学曲线图;(d)为 s-dopa和r-dopa分别作为底物,s-pani(ta)@cu
2+
(蓝色)、 r-pani(ta)@cu
2+
(红色)和pani(ta)@cu
2+
(灰色)作为催化剂下在475nm 处360秒间隔内的吸光度变化图。
具体实施方式
27.下面将结合附图对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
28.此外,下面所描述的本发明不同实施方式中所涉及的技术特征只要彼此之间未构成冲突就可以相互结合。
29.实施例1:超分子手性s-pani(ta)@cu
2+
手性催化剂
30.s1:溶液a为:3.5g s-csa、11.6mg苯胺低聚物和0.2g苯胺单体溶解在1.5ml超纯水,超声溶解均匀,溶液b为:0.49g过硫酸铵溶解在1ml超纯水中,取溶液b 200μl在溶液a搅拌情况下加入,搅拌30s后静置,然后每间隔0.5h加样一次,最后静置24h后超纯水离心洗样3次,得到 s-pani(csa)。
31.s2:将所得s-pani(csa)分散在100ml 0.1mol/l氨水中搅拌3h后,超纯水洗样6次,得到s-pani(undoped)。
32.s3:20mg s-pani(undoped)分散在10ml超纯水中,加入10ml 1m的巯基乙酸水溶液后搅拌3h后,超纯水洗样3次,得到s-pani(ta)。
33.s4:将s-pani(ta)分散在40ml 1
×
10^-4
m的氯化铜水溶液中,搅拌3h 后,超纯水洗样三次后得到超分子手性s-pani(ta)@cu
2+
的手性催化剂。
34.实施例2:
35.同实施例1,区别在于将s-csa替换为r-csa,分别得到r-pani(csa)、r-pani(undoped)、r-pani(ta)和r-pani(ta)@cu
2+
。
36.实施例3:
37.本实施为非手性pani(ta)@cu
2+
催化剂的制备方法
38.s1:1号溶液为:0.6ml苯胺单体溶解在20ml 1m hcl的溶液中,超声均匀,2号溶液为:0.36g过硫酸铵溶解在20ml 1m hcl的溶液中,在1号溶液搅拌情况下将2号溶液加入,搅拌30s后静置24h后超纯水离心洗样3 次,得到pani(csa)。
39.s2:将所得pani(csa)分散在100ml 0.1mol/l氨水中搅拌3h后,超纯水洗样6次,得到pani(undoped)。
40.s3:20mg pani(undoped)分散在10ml超纯水中,加入10ml 1m的巯基乙酸水溶液后搅拌3h后,超纯水洗样3次,得到pani(ta)。
41.表征:
42.通过观察实施例1-3制得的各产物的tem图以及uv和cd光谱图确认产物的手性。
43.一、关于实施例1各产物的手性情况
44.从图1(a1)tem图中可以看到向右螺旋扭曲的形貌,其插图照片中溶液的墨绿色和图2(a)uv谱图表明s-pani(csa)处于掺杂态,图3(d)cd 光谱中可以看到在448nm处有正的较强手性信号峰,表明s-pani(csa)具有手性。从图1(b1)tem图中仍然可以看到向右螺旋扭曲的形貌,表明 s-pani(csa)去掺杂过程后其手性结构基本不受影响,其插图照片中溶液的蓝色和图2(b)uv谱图表明s-pani(undoped)处于未掺杂态,图3(e)cd 光谱中可以看到在365nm处有正的手性信号峰,表明其s-pani(undoped)具有手性。
45.加入巯基乙酸后,从图1(c1)tem图中可以看到向右螺旋扭曲的形貌,其插图照片中溶液的黄绿色和图2(c)uv谱图表明s-pani(ta)处于掺杂态,图3(f)cd光谱中可以看到在458nm处有正的较强手性信号峰,表明其具有手性。
46.吸附铜离子后得到产物s-pani(ta)@cu
2+
的sem图如图4(a1)所示明将其放到得到图5(c1),从图中可以看到明显向右螺旋扭曲的形貌,图6(d1)为tem图,同样可以看到明显
向右螺旋扭曲的形貌,图7(b)cd光谱中可以看到在448nm处有正的较强手性信号峰,并且在396nm处有正的较强新手性信号峰,表明其s-pani(ta)的手性向cu
2+
转移。
47.二、关于实施例2各产物的手性情况
48.图1(a2
→
c2)为实施例2各阶段产物tem图,从图中可以看到向左螺旋扭曲的形貌,图2和图3中亦有说明实施例2各阶段产物的uv谱图和cd 谱图,通过比较发现其与实施例1的性能相近,即r-pani(csa)、 r-pani(undoped)、r-pani(ta)具有手性。
49.图4(a2)、图5(c2)、图6(d2)以及图7表明r-pani(ta)的手性向cu
2+
转移。
50.三、关于实施例3各产物的手性情况
51.图1(a3
→
c3)为实施例3各阶段产物tem图,可以看到纤维状的直线形貌,其插图照片中溶液的墨绿色和图2(a)uv谱图表明pani(csa)处于掺杂态,图3(d)cd光谱中无手性信号峰出现,表明pani(csa)不具有手性。观察tem图和uv、cd谱图,pani(undoped)和pani(ta)同样不具有手性。
52.图4(a3)、图5(c3)、图6(d3)可以看到纤维状的直线形貌,图7的 cd光谱中无手性信号峰出现,表明pani(ta)@cu
2+
不具有手性。
53.应用例
54.s-pani(ta)@cu
2+
,r-pani(ta)@cu
2+
手性催化剂和非手性 pani(ta)@cu
2+
催化剂分别催化r-dopa和s-dopa的动力学研究。
55.动力学测量是通过监测dopa在475nm处随时间的吸光度变化。实验使用100μg ml-1
s-pani(ta)@cu
2+
或r-pani(ta)@cu
2+
或pani(ta)@cu
2+
,反应体积为3ml,缓冲溶液(25mm na2hpo4,ph 7.0,25℃)以200μm dopa 作为底物,h2o2浓度为50mm。
56.从图9(a)中可以看出相同条件下超分子手性s-pani(ta)@cu
2+
对 s-dopa的反应速度比对r-dopa的反应速度要快;从图9(b)中可以看出相同条件下超分子手性r-pani(ta)@cu
2+
对r-dopa的反应速度比对 s-dopa的反应速度要快;从图9(c)中可以看出相同条件下pani(ta)@cu
2+
对s-dopa的反应速度和对s-dopa的反应速度几乎相同。图9(d)中,相同条件下,s-pani(ta)@cu
2+
,r-pani(ta)@cu
2+
手性催化剂和非手性 pani(ta)@cu
2+
催化剂分别催化r-dopa和s-dopa在475nm处360s间隔内的吸光度变化,可以更加明显看出超分子手性s-pani(ta)@cu
2+
对s-dopa 相比较于r-dopa有更强的反应活性,而超分子手性r-pani(ta)@cu
2+
对 r-dopa相比较于s-dopa有更强的反应活性,对比样pani(ta)@cu
2+
对 s-dopa的反应活性和对s-dopa的反应活性几乎相同。超分子手性 s-pani(ta)@cu
2+
对dopa的选择性因子为1.43,表明其对s-dopa的反应快慢是r-dopa的1.43倍,超分子手性r-pani(ta)@cu
2+
对doap的选择性因子为1.44,表明其对r-dopa的反应快慢是s-dopa的1.44倍;而 pani(ta)@cu
2+
对doap的选择性因子为1.01,表明其催化r-dopa和 s-dopa的反应快慢相同。这就能体现出超分子手性pani(ta)@cu
2+
纳米手性催化剂的手性选择性效果。
57.尽管本发明的实施方案已公开如上,但其并不仅仅限于说明书和实施方式中所列运用,它完全可以被适用于各种适合本发明的领域,对于熟悉本领域的人员而言,可容易地实现另外的修改,因此在不背离权利要求及等同范围所限定的一般概念下,本发明并不限于特定的细节和这里示出与描述的图例。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1