一种增强孔隙型Pt基合金膜催化剂及其制备方法
一种增强孔隙型pt基合金膜催化剂及其制备方法
技术领域
1.本发明属于水电解-有机物电催化还原耦合技术领域,具体涉及一种增强孔隙型pt基合金膜催化剂及其制备方法。
背景技术:
2.氢能是一种高效、清洁的能源,目前工业上以通过电解水制氢等方式,大规模制取氢气。但氢气的存储仍是一个难题。液态有机物储氢技术具有储氢密度高、技术成本低以及便于运输等优点,成为目前较为可行的储氢方式。故将水电解制氢技术以及有机物电催化还原技术相耦合,使制氢储氢一体化。该过程具有反应条件温和、储氢效率高的优点,其核心在于膜催化剂,一般选用稳定性好、催化活性高的pt基合金来合成具有高加氢效率的膜催化剂。
3.在pt基催化剂设计制备中,高的几何比表面积(ssa)及电化学活性比表面积(esa)有利于提高其在催化加氢方面的催化效率。目前获得高的ssa、esa的改进,如中国专利cn10924482a,采用化学沉积法制备催化剂,并通过酸蚀法去合金化,可去除催化剂粉体内多余的合金组分并有效提高稳定性和活性,但使用的化学沉积法使催化剂可能含有杂质,且使用单一含氧酸进行酸蚀,无法同时获得高的ssa、esa。如中国专利cn113658810a采用自活化法制备催化剂,具有制备时间短,不需要复杂设备的优点,但获得高的ssa过程复杂,且需要进行基底与催化层分离,可能会造成催化层损失。如中国专利cn113083308b采用浸渍法合成催化剂,具有选择性高,产率高的优点,但制备时间长。
4.因此,亟待提出一种新的方法,同时获得高的ssa、esa以提高其催化活性,并解决制备时间长且过程复杂、贵金属损失较多等问题。
技术实现要素:
5.针对现有技术存在的上述缺陷,本发明提供一种增强孔隙型pt基合金膜催化剂及其制备方法,采用不同阴离子无机酸组合酸蚀,通过控制电化学腐蚀时间、不同阴离子无机酸浓度以及腐蚀温度来调控催化剂表面pt原子迁移率,得到具有高的ssa、esa的孔隙型pt基合金膜催化剂,解决其制备过程复杂、贵金属损失较多等问题。
6.为实现上述目的,本发明提供如下技术方案:
7.技术方案一:一种增强孔隙型pt基合金膜催化剂,以pt为主相元素,过渡金属元素为合金相,稀土元素为催化助剂,在炭质载体上采用真空热沉积的离子束溅射技术制备pt基合金膜催化剂,然后采用无机酸对所得pt基合金膜催化剂进行两次电化学腐蚀,即得到增强孔隙型pt基合金膜催化剂。
8.进一步地,所述过渡金属元素为ti、ni或cu中的一种;所述稀土元素包括ce或la。
9.进一步地,所述炭质载体为石墨纤维布、炭纸或石墨片中的一种。
10.进一步地,所述无机酸为不含氧无机酸或含氧无机酸。
11.进一步地,所述不含氧无机酸为hcl或hbr,所述含氧无机酸为hclo4或h2so4。
12.进一步地,所述不含氧无机酸浓度为0.5-1.0mol/l;含氧无机酸浓度为0.25-0.75mol/l。
13.所述两次电化学腐蚀具体是指:先进行不含氧无机酸电化学腐蚀,再经室温去离子水冲洗后,进行含氧无机酸电化学腐蚀;或先进行含氧无机酸电化学腐蚀,再经室温去离子水冲洗后,进行不含氧无机酸电化学腐蚀。
14.进一步地,所述电化学腐蚀温度为30-60℃,腐蚀时间为5-60min。
15.进一步地,所述增强孔隙型pt基合金膜催化剂几何比表面积为81.1-265m2/g,电化学活性比表面积为734-1183m2/g。
16.技术方案二:一种所述的增强孔隙型pt基合金膜催化剂的制备方法,包括以下步骤:
17.1)将炭质载体浸没在1.0mol/l的h2so4溶液中,超声清洗8min后使用去离子水冲洗,再放入丙酮溶液中超声清洗15min后使用去离子水冲洗,再进行45min干燥脱水处理,得到预处理的炭质载体;
18.2)将步骤1)所得预处理的炭质载体放置在离子束溅射装置的样品台上,再将pt靶、过渡金属靶以及稀土靶安装在离子束溅射装置的靶台上,抽真空至8.0x10-4
pa,并在真空度达到2.0x10-3
pa时,开始加热样品台至200℃-350℃后使用离子束辅助溅射装置清洗6min,再使用离子束溅射靶材制备pt基合金膜催化剂。
19.贵金属pt在液态有机物储氢方面具有良好的催化性能,但其价格昂贵,纯pt作为阴极材料易发生co中毒失活,而第二元素的加入不仅可以降低其pt载量,而且可以提高其催化活性以及稳定性。本发明在pt中掺杂过渡金属元素,如ti的电负性低于pt的电负性,电子可能从ti转移到pt上从而增加pt的电子云密度。稀土催化助剂如ce,与pt有良好的协同作用,因此构成的三元合金催化剂可以提高pt的co中毒抗性、催化活性以及降低pt载量。
20.与现有技术相比,本发明的有益效果为:
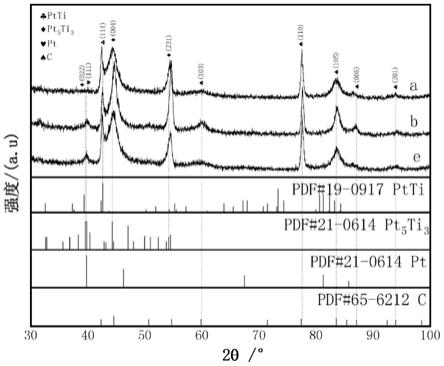
21.1.本发明采用离子束溅射沉积,真空热处理技术以及不同阴离子无机酸两步酸蚀法进行电化学组合腐蚀,制备增强孔隙型pt基合金膜催化剂。
22.2.本发明采用的两步酸蚀法可依次控制pt基合金膜催化剂表面的pt原子迁移率,以获得具有高的ssa、esa的增强孔隙型pt基合金膜催化剂,增加了膜催化剂与接触液的反应面积,同时可以调控参与析氢反应的活性位点数量,提高了析氢催化性能。从而直接应用于水电解-有机物电催化还原耦合技术领域。
23.3.本发明采用不同阴离子无机酸进行电化学修饰。无机不含氧酸(如hcl)中的h
+
浓度较高且cl-能够极大增强ce和ti原子的扩散速率,从而提高催化剂的ssa;而无机含氧酸(如hclo4)能够调控参与析氢反应的活性位点数量,从而提高催化剂的esa,增加催化剂的析氢反应速率。
24.4.本发明采用的炭质载体,可使用离子束辅助清洗处理,可增强pt基合金催化剂膜层与炭质载体的结合强度及炭质载体的导电性,从而提高增强孔隙型pt基合金膜催化剂的电催化活性。
25.5.本发明的制备过程简便,无中间污染物。
附图说明
26.为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
27.图1为对照组、实施例6和实施例4制备的孔隙型pt-ti-ce合金膜催化剂的xrd叠加图谱(30
°
≤2θ≤100
°
),其中a、b和e分别代表对照组、实施例6和实施例4的xrd图谱;
28.图2为对照组、实施例6、实施例8、实施例2以及实施例4制备的孔隙型pt-ti-ce合金膜催化剂的cv曲线对比图,其中a、b、c、d以及e分别代表对照组、实施例6、实施例8、实施例2以及实施例4的cv曲线;插图为孔隙型pt-ti-ce合金膜催化剂的cv曲线局部放大图,图中s1、s2、s3、s4以及s5分别为对照组、实施例6、实施例8、实施例2以及实施例4的氢的脱附峰的积分面积;
29.图3是对照组、实施例6、实施例8、实施例2以及实施例4制备的孔隙型pt-ti-ce合金膜催化剂的lsv曲线对比图,其中a、b、c、d以及e分别代表对照组、实施例6、实施例8、实施例2以及实施例4的lsv曲线;
30.图4是对照组制备的pt-ti-ce合金膜催化剂表面的stem照片;
31.图5是实施例6制备的孔隙型pt-ti-ce合金膜催化剂表面的stem照片;
32.图6是实施例4制备的孔隙型pt-ti-ce合金膜催化剂表面的stem照片。
具体实施方式
33.现详细说明本发明的多种示例性实施方式,该详细说明不应认为是对本发明的限制,而应理解为是对本发明的某些方面、特性和实施方案的更详细的描述。
34.应理解本发明中所述的术语仅仅是为描述特别的实施方式,并非用于限制本发明。另外,对于本发明中的数值范围,应理解为还具体公开了该范围的上限和下限之间的每个中间值。在任何陈述值或陈述范围内的中间值以及任何其他陈述值或在所述范围内的中间值之间的每个较小的范围也包括在本发明内。这些较小范围的上限和下限可独立地包括或排除在范围内。
35.除非另有说明,否则本文使用的所有技术和科学术语具有本发明所述领域的常规技术人员通常理解的相同含义。虽然本发明仅描述了优选的方法和材料,但是在本发明的实施或测试中也可以使用与本文所述相似或等同的任何方法和材料。本说明书中提到的所有文献通过引用并入,用以公开和描述与所述文献相关的方法和/或材料。在与任何并入的文献冲突时,以本说明书的内容为准。
36.在不背离本发明的范围或精神的情况下,可对本发明说明书的具体实施方式做多种改进和变化,这对本领域技术人员而言是显而易见的。由本发明的说明书得到的其他实施方式对技术人员而言是显而易见的。本技术说明书和实施例仅是示例性的。
37.关于本文中所使用的“包含”、“包括”、“具有”、“含有”等等,均为开放性的用语,即意指包含但不限于。
38.本发明各实施例制备的孔隙型pt基合金膜催化剂可使用x射线衍射仪(xrd)、电感耦合等离子体光谱分析仪(icp-oes)、氮吸附比表面积分析仪(bet)以及扫描透射电镜
(stem)等来评价其物相组成、元素含量、ssa以及表面形貌。
39.本发明制备的孔隙型pt基合金膜催化剂可使用配合电化学工作站的三电极密封电解池体系,采用循环伏安法(cv)以及线性扫描伏安法(lsv)等来评价其esa以及交换电流密度(i0)。
40.其中:测试溶液为去除溶解氧后30℃的0.5mol/l h2so4溶液,cv测试电势扫描范围为-0.35~1.2v(相对饱和甘汞电极),扫描速度为50mv/s。根据cv曲线中氢的脱附峰的积分面积(直接反映表面活性反应位数量),可以得到单位质量pt的esa。见公式(1):
[0041][0042]
式中:esa-单位质量pt的电化学活性比表面积;s-氢的脱附峰的积分面积;m-1cm2工作电极上pt的含量;v-扫描速率;c-pt对氢的单位吸附电容(0.21mc/cm2)
[0043]
lsv测试扫描区间为-0.40~-0.28v(相对饱和甘汞电极),扫描速率为50mv/s,并通过公式(2)获得i0以评价膜电极的催化效率。
[0044]
lga=kδe+lgi0ꢀꢀ
(2)
[0045]
其中
[0046][0047]
式中:k-常数;δe-超电势;i
0-交换电流密度;f-法拉第常数;r-气体常数;t-电极反应温度;z-电荷数。
[0048]
实施例1
[0049]
1)将尺寸为100
×
100mm2的石墨纤维布浸没在室温的1.0mol/l的h2so4溶液中超声清洗8min后使用去离子水冲洗,再放入丙酮溶液中超声清洗15min后使用去离子水冲洗,再进行45min干燥脱水处理,得到预处理的石墨纤维布;
[0050]
2)将步骤1)所得石墨纤维布放置在离子束溅射装置的样品台上,再将pt靶、ti靶以及ce靶安装在离子束溅射装置的靶台上,抽真空至8.0x10-4
pa,并在真空度达到2.0x10-3
pa时,开始加热样品台至350℃。在温度为350℃、8.0
×
10-4
pa的真空中经离子束辅助清洗6min后,通入6sccm的高纯ar,控制溅射屏压为2kv、束流为60ma,产生离子束溅射pt靶、ti靶以及ce靶(50wt%:20wt%:30wt%)15min,再在相同真空度内经自然冷却至室温后得到pt-ti-ce合金膜催化剂;
[0051]
3)将步骤2)制备的pt-ti-ce合金膜催化剂裁成15
×
15mm2的试样若干,任取四份分别先放入50℃的0.1mol/l hcl溶液中酸蚀30min,酸蚀完毕后用室温去离子水冲洗,再放入60℃的0.75mol/l hclo4溶液中酸蚀10min,将酸蚀后的样品使用室温去离子水冲洗,即制得孔隙型pt-ti-ce合金膜催化剂。
[0052]
对得到的孔隙型pt-ti-ce合金膜催化剂进行bet测试、cv测试、lsv测试以及icp测试,获得其ssa、esa、i0以及元素含量。
[0053]
结果:本实施例孔隙型pt-ti-ce合金膜催化剂的ssa为265m2/g,较对照组提升472%;esa为1152m2/g,较对照组提升180%;单位ssa对esa增加的有效率为38%;i0为3.223ma/cm2;pt含量为0.061mg/cm2。
[0054]
实施例2
[0055]
同实施例1,区别在于,步骤3)具体操作为:将步骤2)制备的pt-ti-ce合金膜催化剂裁成15
×
15mm2的试样若干,任取四份分别先放入50℃的0.1mol/l hcl溶液中酸蚀30min,酸蚀完毕后用室温去离子水冲洗,再放入60℃的0.75mol/l hclo4溶液中酸蚀5min,将酸蚀后的样品使用室温去离子水冲洗,即制得孔隙型pt-ti-ce合金膜催化剂。
[0056]
对得到的孔隙型pt-ti-ce合金膜催化剂进行bet测试、cv测试、lsv测试以及icp测试,获得其ssa、esa、i0以及元素含量。
[0057]
结果:本实施例孔隙型pt-ti-ce合金膜催化剂的ssa为242.913m2/g,较对照组提升433%;esa为1163m2/g,较对照组提升181%;单位ssa对esa增加的有效率为41%;i0为3.817ma/cm2;pt含量为0.065mg/cm2。
[0058]
实施例3
[0059]
同实施例1,区别在于,步骤3)具体操作为:将步骤2)制备的pt-ti-ce合金膜催化剂裁成15
×
15mm2的试样若干,任取四份分别先放入60℃的0.75mol/l hclo4溶液中酸蚀20min,酸蚀完毕后用室温去离子水冲洗,再放入50℃的1.0mol/l hcl溶液中酸蚀8min,将酸蚀后的样品使用室温去离子水冲洗,即制得孔隙型pt-ti-ce合金膜催化剂。
[0060]
对得到的孔隙型pt-ti-ce合金膜催化剂进行bet测试、cv测试、lsv测试以及icp测试,获得其ssa、esa、i0以及元素含量。
[0061]
结果:本实施例pt-ti-ce合金膜催化剂的ssa为140m2/g,较对照组提升249%;esa为1019m2/g,较对照组提升159%;单位ssa对esa增加的有效率为63%;i0为3.798ma/cm2;pt含量为0.086mg/cm2。
[0062]
实施例4
[0063]
同实施例3,区别在于,步骤3)中hcl的浓度为0.1mol/l。
[0064]
对得到的孔隙型pt-ti-ce合金膜催化剂进行bet测试、cv测试、lsv测试以及icp测试,获得其ssa、esa、i0以及元素含量。
[0065]
结果:本实施例孔隙型pt-ti-ce合金膜催化剂的xrd图中存在ptti(111)、pt5ti3(022)以及pt(111)的特征衍射峰;ssa为196.35m2/g,较对照组提升350%;esa为1183m2/g,较对照组提升184%;单位ssa对esa增加的有效率为52%;i0为3.803ma/cm2;pt含量为0.0750mg/cm2。
[0066]
实施例5
[0067]
同实施例1,区别在于,步骤3)具体操作为:将步骤2)制备的pt-ti-ce合金膜催化剂裁成15
×
15mm2的试样若干,任取四份分别先放入40℃的0.75mol/l hcl溶液中酸蚀60min,将酸蚀后的样品使用室温去离子水冲洗,即制得孔隙型pt-ti-ce合金膜催化剂。
[0068]
对得到的孔隙型pt-ti-ce合金膜催化剂进行bet测试、cv测试、lsv测试以及icp测试,获得其ssa、esa、i0以及元素含量。
[0069]
结果:本实施例孔隙型pt-ti-ce合金膜催化剂的ssa为196.3m2/g,较对照组提升349%;esa为967m2/g,较对照组提升151%;单位ssa对esa增加的有效率为43%;i0为3.376ma/cm2;pt含量为0.059mg/cm2。
[0070]
实施例6
[0071]
同实施例1,区别在于,步骤3)具体操作为:将步骤2)制备的pt-ti-ce合金膜催化
剂裁成15
×
15mm2的试样若干,任取四份分别先放入50℃的1.0mol/l hcl溶液中酸蚀30min,将酸蚀后的样品使用室温去离子水冲洗,即制得孔隙型pt-ti-ce合金膜催化剂。
[0072]
对得到的孔隙型pt-ti-ce合金膜催化剂进行bet测试、cv测试、lsv测试以及icp测试,获得其ssa、esa、i0以及元素含量。
[0073]
结果:本实施例孔隙型pt-ti-ce合金膜催化剂的xrd图中存在ptti(111)、pt5ti3(022)以及pt(111)的特征衍射峰;ssa为222.1m2/g,较对照组提升395%;esa为1180m2/g,较对照组提升184%;单位ssa对esa增加的有效率为46%;i0为3.747ma/cm2;pt含量为0.069mg/cm2。
[0074]
实施例7
[0075]
同实施例1,区别在于,步骤3)具体操作为:将步骤2)制备的pt-ti-ce合金膜催化剂裁成15
×
15mm2的试样若干,任取四份分别先放入50℃的0.5mol/l hclo4溶液中酸蚀60min,将酸蚀后的样品使用室温去离子水冲洗,即制得孔隙型pt-ti-ce合金膜催化剂。
[0076]
对得到的孔隙型pt-ti-ce合金膜催化剂进行bet测试、cv测试、lsv测试以及icp测试,获得其ssa、esa、i0以及元素含量。
[0077]
结果:本实施例孔隙型pt-ti-ce合金膜催化剂的ssa为81.1m2/g,较对照组提升144%;esa为734m2/g,较对照组提升114%;单位ssa对esa增加的有效率为79%;i0为3.509ma/cm2;pt含量为0.102mg/cm2。
[0078]
实施例8
[0079]
同实施例1,区别在于,步骤3)具体操作为:将步骤2)制备的pt-ti-ce合金膜催化剂裁成15
×
15mm2的试样若干,任取四份分别先放入60℃的0.75mol/l hclo4溶液中酸蚀20min,将酸蚀后的样品使用室温去离子水冲洗,即制得孔隙型pt-ti-ce合金膜催化剂。
[0080]
对得到的孔隙型pt-ti-ce合金膜催化剂进行bet测试、cv测试、lsv测试以及icp测试,获得其ssa、esa、i0以及元素含量。
[0081]
结果:本实施例孔隙型pt-ti-ce合金膜催化剂的ssa为86m2/g,较对照组提升153%;esa为770m2/g,较对照组提升120%;单位ssa对esa增加的有效率为78%;i0为3.577ma/cm2;pt含量为0.098mg/cm2。
[0082]
对照组1
[0083]
同实施例1,区别在于,步骤3)具体操作为:将步骤2)制备的pt-ti-ce合金膜催化剂裁成15
×
15mm2的试样若干,任取四份分别进行bet测试、cv测试、lsv测试以及icp测试,获得其ssa、esa、i0以及元素含量。
[0084]
结果:本对照组pt-ti-ce合金膜催化剂的xrd图中存在ptti(111)、pt5ti3(022)以及pt(111)的特征衍射峰,ssa为56.1m2/g,esa为640m2/g,i0为2.997ma/cm2,pt含量为0.115mg/cm2。
[0085]
图1~6为对照组及实施例的xrd、cv、lsv以及stem测试结果:
[0086]
图1中a、b、e分别为对照组、实施例6以及实施例4的xrd谱线。出现ptti(111)、pt5ti3(022)、pt5ti3(004)、pt5ti3(231)以及pt(111)的特征衍射峰,表明催化剂纯净无杂质元素。图1a中,出现ptti(111)、pt5ti3(022)、pt5ti3(004)以及pt5ti3(231)的特征衍射峰,表明在热处理时,ptti形成了合金相,且pt单质与ti单质的含量很少。图1中b、e谱线与a谱线相比,出现pt(111)峰,表明大量ti被腐蚀,出现了富pt相。
[0087]
图2中a、b、c、d以及e分别为对照组、实施例6、实施例8、实施例2以及实施例4的cv曲线。图2中插图为孔隙型pt-ti-ce合金膜催化剂的cv曲线局部放大图,其中s1、s2、s3、s4以及s5分别为对照组、实施例6、实施例8、实施例2以及实施例4的氢的脱附峰的积分面积。使用公式(1)得出对照组、实施例6、实施例8、实施例2以及实施例4的esa值,分别为640m2/g、1180m2/g、770m2/g、1163m2/g以及1183m2/g,表明通过不同阴离子无机酸组合酸蚀可获得高的esa。对照组的析氢性能弱于实施例2、实施例4,由于不同阴离子无机酸组合蚀刻ti,使活性位点数增加,提高其催化性能。
[0088]
图3中a、b、c、d以及e分别为对照组、实施例6、实施例8、实施例2以及实施例4的lsv曲线。利用公式(2)得出对照组、实施例6、实施例8、实施例2以及实施例4的i0分别为2.997ma/cm2、3.747ma/cm2、3.577ma/cm2、3.817ma/cm2以及3.803ma/cm2,表明通过不同阴离子无机酸组合酸蚀,可以提高孔隙型pt-ti-ce合金膜催化剂的析氢性能。
[0089]
图4为对照组的stem照片,可以看出其膜层凹凸不平且有极少数孔洞。
[0090]
图5为实施例6制备的孔隙型pt-ti-ce合金膜催化剂的stem照片。其表面成火山结构,且出现直径<10nm的孔洞。
[0091]
图6为实施例4制备的孔隙型pt-ti-ce合金膜催化剂的stem照片。其表面为孔隙性结构,且与实施例6相比孔径大大增加,为100nm左右,主要以ptti合金为主体,并出现富pt相。
[0092]
实施例9
[0093]
同实施例4,区别在于,将ti替换成ni,得到孔隙型pt-ni-ce合金膜催化剂。
[0094]
结果:本实施例孔隙型pt-ni-ce合金膜催化剂的ssa为172m2/g,较对照组提升306%;esa为973m2/g,较对照组提升152%;单位ssa对esa增加的有效率为49%;i0为3.753ma/cm2;pt含量为0.0695mg/cm2。
[0095]
实施例10
[0096]
同实施例4,区别在于,将ti替换成cu,得到孔隙型pt-cu-ce合金膜催化剂。
[0097]
结果:本实施例孔隙型pt-cu-ce合金膜催化剂的ssa为169.1m2/g,较对照组提升300%;esa为1103m2/g,较对照组提升172%;单位ssa对esa增加的有效率为57%;i0为3.752ma/cm2;pt含量为0.0737mg/cm2。
[0098]
实施例11
[0099]
同实施例4,区别在于,将石墨纤维布替换成炭纸,得到孔隙型pt-ti-ce合金膜催化剂。
[0100]
结果:本实施例孔隙型pt-ti-ce合金膜催化剂的ssa为183.42m2/g,较对照组提升326%;esa为1172m2/g,较对照组提升183%;单位ssa对esa增加的有效率为56%;i0为3.679ma/cm2;pt含量为0.0784mg/cm2。
[0101]
实施例12
[0102]
同实施例4,区别在于,步骤3)中,含氧无机酸替换为h2so4,不含氧无机酸替换为hbr,得到孔隙型pt-ti-ce合金膜催化剂。
[0103]
结果:本实施例孔隙型pt-ti-ce合金膜催化剂的ssa为189.1m2/g,较对照组提升337%;esa为1083m2/g,较对照组提升169%;单位ssa对esa增加的有效率为50%;i0为3.631ma/cm2;pt含量为0.0642mg/cm2。
[0104]
以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1