一种卤氧化铋固溶体光电薄膜、其制备方法及应用与流程
1.本发明涉及固溶体光电薄膜技术领域,尤其涉及一种卤氧化铋固溶体光电薄膜、其制备方法及应用。
背景技术:
2.工业废水的综合治理已成为我国亟待解决的重大环境问题。2018年,我国工业废水排放量达187亿吨,其中造纸、皮革、农药、染料等行业排出的废水含有高浓度的难降解有机物,是工业废水的主要污染源。因难降解有机物具有抗光解、抗热及抗生物等性质,传统的物理吸附、混凝、沉降等方法无法实现有机污染物的有效降解,以光催化/光电催化降解为代表的高级氧化法(aops)技术应运而生。光催化/光电催化降解技术能在自然光的激发下原位产生具有高氧化能力的空穴、羟基自由基、超氧自由基等活性物质,进而实现有机染料、氯化有机物、酚类化合物、杀虫剂等有机污染物的有效降解,具有无污染、高能效、低成本的优点,因此被认为是最具发展潜力的有机废水处理方法之一。
3.卤氧化铋(biox,x=f,cl,br,i)具有pbfcl型晶体结构,由以[x-bi-o-bi-x]为基本单元的层状结构通过卤素原子间的弱范德华力沿[001]方向堆垛而成,带正电的铋氧层[bi2o2]
2+
和带负电的卤素离子层(x-)在[001]方向诱导生成内电场,可有效促进光生载流子的分离,因其独特的层状结构、适宜的带隙宽度及优异的催化活性,biox受到研究者的广泛关注,是一种具有可见光响应的新型光催化/光电催化材料,被广泛应用于光催化/光电催化、光电传感器、太阳能电池等领域。
[0004]
通常,利用水热法、溶剂热法、共沉淀法制备出的biox是以纳米粉体形式存在,但在处理有机废水的实际应用中,纳米粉体材料存在诸多不足,如在运输、装填、排放过程中存在粉尘污染,不利于人工操作、在使用过程中容易出现淤积,堵塞管路、不易从溶液中分离,易造成二次污染等,严重局限了biox的实际应用。
技术实现要素:
[0005]
有鉴于此,本发明要解决的技术问题在于提供一种卤氧化铋固溶体光电薄膜、其制备方法及应用,可以避免产生上述问题,制备的卤氧化铋固溶体光电薄膜可用于有机污染物的光催化/光电催化降解、高附加值化工产品的光催化合成以及制备光电催化产氢电极。
[0006]
本发明提供了一种卤氧化铋固溶体光电薄膜的制备方法,包括以下步骤:
[0007]
a)将含铋化合物的水溶液和卤盐混匀后,得到第一混合溶液;
[0008]
b)将对苯醌的乙醇溶液滴加至所述第一混合溶液中,混匀后,得到第二混合溶液;
[0009]
c)调整所述第二混合溶液的ph值为0.6~4.0,得到电化学沉积液;
[0010]
d)采用所述电化学沉积液进行电化学沉积,得到薄膜前体;
[0011]
e)将所述薄膜前体进行退火处理,得到卤氧化铋固溶体光电薄膜。
[0012]
优选的,步骤a)中,所述含铋化合物包括硝酸铋、氢氧化铋、碘化铋、溴化铋或氧化
铋;
[0013]
所述含铋化合物的水溶液的浓度为10mmol/l~40mmol/l。
[0014]
优选的,步骤a)中,所述卤盐包括碘化钠、溴化钠、碘化钾和溴化钾中的一种或两种;
[0015]
所述第一混合溶液中卤盐的摩尔浓度为0.4~2.0mol/l。
[0016]
优选的,步骤b)中,所述对苯醌的乙醇溶液的浓度为0.1~0.4mol/l。
[0017]
优选的,步骤b)中,所述混匀的温度为10~30℃。
[0018]
优选的,步骤c)中,调整所述第二混合溶液的ph值采用浓硝酸、硫酸或盐酸。
[0019]
优选的,步骤d)中,所述电化学沉积采用三电极体系,包括参比电极、对电极以及作为沉积基底的工作电极;
[0020]
电化学沉积过程中采用恒电位法;
[0021]
所述电化学沉积的沉积电位为-0.3~0.3v
ag/agcl
,沉积时间为20~900s。
[0022]
优选的,步骤e)中,所述退火处理的温度为100~300℃,时间为1~2h。
[0023]
本发明还提供了一种上文所述的制备方法制得的卤氧化铋固溶体光电薄膜。
[0024]
本发明还提供了一种上文所述的卤氧化铋固溶体光电薄膜在光催化降解、电催化降解、光电催化降解、光催化合成或者在制备光电催化产氢电极方面的应用。
[0025]
本发明提供了一种卤氧化铋固溶体光电薄膜的制备方法,包括以下步骤:a)将含铋化合物的水溶液和卤盐混匀后,得到第一混合溶液;b)将对苯醌的乙醇溶液滴加至所述第一混合溶液中,混匀后,得到第二混合溶液;c)调整所述第二混合溶液的ph值为0.6~4.0,得到电化学沉积液;d)采用所述电化学沉积液进行电化学沉积,得到薄膜前体;e)将所述薄膜前体进行退火处理,得到卤氧化铋固溶体光电薄膜。本发明制备的卤氧化铋固溶体光电薄膜不仅具有可调的卤素比例,而且还能实现薄膜颜色、光学带隙及微观形貌的调控,可用于有机污染物的光催化降解、电催化降解、光电催化降解、高附加值化工产品的光催化合成以及制备光电催化产氢电极。因此,本发明的卤氧化铋固溶体光电薄膜有利于为光电薄膜材料开拓出更加广泛的应用范围。
附图说明
[0026]
图1为本发明实施例中样品s1和样品s6的xrd图谱以及对应的标准pdf卡片;
[0027]
图2为本发明实施例中样品s1~s6的xrd图谱;
[0028]
图3为本发明实施例中样品s1~s6的eds元素分析对比图;
[0029]
图4为本发明实施例中样品s1~s6的颜色对比照片;
[0030]
图5为本发明实施例中样品s1~s6的紫外可见光吸收光谱图;
[0031]
图6为实施例中样品s1~s6的tauc图;
[0032]
图7为本发明实施例中样品s1~s6的sem图;
[0033]
图8为本发明实施例中样品s1~s6的莫特肖特基曲线图;
[0034]
图9为本发明实施例中样品s1~s6的能带结构示意图;
[0035]
图10为本发明实施例中样品s1退火处理前后的i-v曲线图;
[0036]
图11为本发明实施例中样品s5的光催化、电催化及光电催化降解rhb的降解性能;
[0037]
图12为本发明实施例中样品s5的光催化、电催化及光电催化降解rhb的降解动力
学曲线。
具体实施方式
[0038]
下面将结合本发明实施例,对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0039]
本发明提供了一种卤氧化铋固溶体光电薄膜的制备方法,包括以下步骤:
[0040]
a)将含铋化合物的水溶液和卤盐混匀后,得到第一混合溶液;
[0041]
b)将对苯醌的乙醇溶液滴加至所述第一混合溶液中,混匀后,得到第二混合溶液;
[0042]
c)调整所述第二混合溶液的ph值为0.6~4.0,得到电化学沉积液;
[0043]
d)采用所述电化学沉积液进行电化学沉积,得到薄膜前体;
[0044]
e)将所述薄膜前体进行退火处理,得到卤氧化铋固溶体光电薄膜。
[0045]
本发明先将含铋化合物的水溶液和卤盐混匀后,得到第一混合溶液。
[0046]
在本发明的某些实施例中,所述含铋化合物包括硝酸铋、氢氧化铋、碘化铋、溴化铋或氧化铋。所述含铋化合物的水溶液的浓度为10mmol/l~40mmol/l。在某些实施例中,所述含铋化合物的水溶液的浓度为20mmol/l。
[0047]
在本发明的某些实施例中,所述含铋化合物的水溶液按照以下方法进行制备:
[0048]
将含铋化合物与水混合,搅拌至完全溶解后,超声分散均匀,得到含铋化合物的水溶液。
[0049]
在本发明的某些实施例中,所述卤盐包括碘化钠、溴化钠、碘化钾和溴化钾中的一种或两种。在某些实施例中,所述卤盐为碘化钠或溴化钠。在某些实施例中,所述卤盐为碘化钠和溴化钠,所述碘化钠和溴化钠的摩尔比为1~4:1~4。在某些实施例中,所述碘化钠和溴化钠的摩尔比为4:1、3:2、2:3或1:4。
[0050]
在本发明的某些实施例中,将含铋化合物的水溶液和卤盐混匀包括:
[0051]
将含铋化合物的水溶液和卤盐混合后,超声分散均匀,得到第一混合溶液。
[0052]
在本发明的某些实施例中,所述第一混合溶液中卤盐的摩尔浓度为0.4~2.0mol/l。在某些实施例中,所述第一混合溶液中卤盐的摩尔浓度为1mol/l。
[0053]
得到第一混合溶液后,将对苯醌的乙醇溶液滴加至所述第一混合溶液中,混匀后,得到第二混合溶液。
[0054]
在本发明的某些实施例中,所述滴加为逐滴滴加。
[0055]
在本发明的某些实施例中,所述对苯醌的乙醇溶液按照以下方法制备:
[0056]
将无水乙醇和对苯醌混合,搅拌至完全溶解后,超声分散均匀,得到对苯醌的乙醇溶液。
[0057]
在本发明的某些实施例中,所述对苯醌的乙醇溶液的浓度为0.1~0.4mol/l。在某些实施例中,所述对苯醌的乙醇溶液的浓度为0.3mol/l。
[0058]
在本发明的某些实施例中,所述混匀的温度为10~30℃。在某些实施例中,所述混匀在室温下进行。在某些实施例中,所述混匀的时间为20~40min。在某些实施例中,所述混匀的时间为30min。在本发明的某些实施例中,所述混匀在磁力搅拌条件下进行。
[0059]
得到第二混合溶液后,调整所述第二混合溶液的ph值为0.6~4.0,得到电化学沉积液。
[0060]
在本发明的某些实施例中,调整所述第二混合溶液的ph值采用浓硝酸、硫酸或盐酸。在某些实施例中,所述浓硝酸的质量浓度为68%。
[0061]
具体的,包括:
[0062]
在搅拌的第二混合溶液中滴加浓硝酸,调节ph值为0.6~4.0,得到电化学沉积液。
[0063]
在本发明的某些实施例中,调整所述第二混合溶液的ph值为4.0、3.8、3.4、2.8或2.0。
[0064]
酸溶液可抑制铋离子的水解,从而提高电化学沉积薄膜的均匀性。
[0065]
得到电化学沉积液,采用所述电化学沉积液进行电化学沉积,得到薄膜前体。
[0066]
在本发明的某些实施例中,所述电化学沉积采用三电极体系,包括参比电极、对电极以及作为沉积基底的工作电极;以ag/agcl为参比电极,pt片为对电极,fto为工作电极。
[0067]
在本发明的某些实施例中,电化学沉积过程中采用恒电位法。
[0068]
在本发明的某些实施例中,所述电化学沉积的沉积电位为-0.3~0.3v
ag/agcl
,沉积时间为20~900s。在某些实施例中,所述电化学沉积的沉积电位为-0.1v
ag/agcl
,沉积时间为300s。
[0069]
在本发明的某些实施例中,所述电化学沉积完成后,还包括:
[0070]
依次用去离子水和乙醇冲洗,然后干燥。
[0071]
本发明对所述冲洗和干燥的方法和参数并无特殊的限制,采用本领域技术人员熟知的冲洗和干燥的方法和参数即可。
[0072]
得到薄膜前体后,将所述薄膜前体进行退火处理,得到卤氧化铋固溶体光电薄膜。
[0073]
在本发明的某些实施例中,所述退火处理的温度为100~300℃,时间为1~2h。在某些实施例中,所述退火处理的温度为250℃,时间为1h。
[0074]
本发明对上文采用的原料来源并无特殊的限制,可以为一般市售。
[0075]
本发明中,所述第一混合溶液中卤盐的摩尔浓度不小于0.4mol/l,高浓度卤素离子与铋离子之间的配位作用,可提高铋离子的溶解度,从而提高电化学沉积薄膜的均匀性。
[0076]
本发明中,通过将至少一种及以上的卤素离子源进行混合,制备卤素离子源溶液,并通过电化学沉积方法制备卤氧化铋固溶体光电薄膜,其中卤氧化铋固溶体光电薄膜的卤素比例可通过卤素源溶液中的卤素比例进行调节;
[0077]
本发明中,通过改变上述至少一种卤氧化铋固溶体光电薄膜的卤素比例,不仅可以实现薄膜颜色、光学带隙在一定范围内可变,还可对光电薄膜的微观形貌进行调控。
[0078]
本发明中,通过改变卤氧化铋固溶体光电薄膜的退火温度及时长,可以改变卤氧化铋固溶体光电薄膜的光电性能。实验表明,卤氧化铋固溶体光电薄膜的结晶度及卤素空位受到退火条件的影响,进而产生不同的光电性能。
[0079]
本发明中,通过改变卤氧化铋固溶体光电薄膜的沉积电位,可以改变卤氧化铋固溶体光电薄膜的光电性能。实验表明,卤氧化铋固溶体光电薄膜的晶面取向易于受到沉积电位的影响,进而产生不同的光电性能。
[0080]
本发明还提供了一种上文所述的制备方法制得的卤氧化铋固溶体光电薄膜。
[0081]
在本发明的某些实施例中,所述卤氧化铋固溶体光电薄膜可以是biobr
xi1-x
(0≤x
≤1)、biocl
xi1-x
(0≤x≤1)、biocl
x
br
1-x
(0≤x≤1)、biocl
x
br
yi1-x-y
(0≤x≤1,0≤y≤1,0≤1-x-y≤1)中的一种。在某些实施例中,所述卤氧化铋固溶体光电薄膜可以是biobr
xi1-x
(0≤x≤1),其中,x=0、0.1、0.2、0.3、0.4、0.5、0.6、0.7、0.8、0.9或1。
[0082]
本发明提供的卤氧化铋固溶体光电薄膜的外观颜色在橙红色至灰白色之间可调,光吸收带边的范围在637nm至360nm之间可调,光学带隙在1.8ev至3.5ev之间可调。
[0083]
本发明还提供了一种上文所述的卤氧化铋固溶体光电薄膜在光催化降解、电催化降解、光电催化降解、光催化合成或者在制备光电催化产氢电极方面的应用;具体的,可以为在光催化降解有机污染物、电催化降解有机污染物、光电催化降解有机污染物、光催化合成化工产品或者在制备光电催化产氢电极方面的应用。在本发明的某些实施例中,所述有机污染物可以为罗丹明b。
[0084]
为了进一步说明本发明,以下结合实施例对本发明提供的一种卤氧化铋固溶体光电薄膜、其制备方法及应用进行详细描述,但不能将其理解为对本发明保护范围的限定。
[0085]
实施例1
[0086]
1、向硝酸铋中加水,搅拌至完全溶解,超声分散均匀后,制得20mmol/l的硝酸铋水溶液;量取100ml上述硝酸铋水溶液,并加入碘化钠,超声分散均匀,得到第一混合溶液;所述第一混合溶液中碘化钠的摩尔浓度为1mol/l;
[0087]
2、将无水乙醇和对苯醌混合,搅拌至完全溶解,超声分散均匀后,制得0.3mol/l的对苯醌乙醇溶液;
[0088]
3、在磁力搅拌条件下,将对苯醌乙醇溶液逐滴滴加到第一混合溶液中,室温下搅拌30min后,在搅拌的第二混合溶液中滴加质量浓度为68%的浓硝酸,调节ph值为4.0,得到电化学沉积液;
[0089]
4、采用所述电化学沉积液进行电化学沉积;
[0090]
所述电化学沉积采用三电极体系,以ag/agcl为参比电极,pt片为对电极,fto为工作电极,以所述电化学沉积液为电解液;在沉积过程采用恒电位法;所述电化学沉积的沉积电位为-0.1v
ag/agcl
,沉积时间为300s;
[0091]
电化学沉积后,将所述薄膜前体依次用去离子水和乙醇冲洗,干燥后薄膜前体;
[0092]
5、将所述薄膜前体在250℃下退火处理1h,得到卤氧化铋固溶体光电薄膜biobr
xi1-x
(x=0),命名为s1。
[0093]
实施例2
[0094]
1、向硝酸铋中加水,搅拌至完全溶解,超声分散均匀后,制得20mmol/l的硝酸铋水溶液;量取100ml上述硝酸铋水溶液,并加入碘化钠和溴化钠(碘化钠和溴化钠的摩尔比为4:1),超声分散均匀,得到第一混合溶液;所述第一混合溶液中卤化钠(包括碘化钠和溴化钠)的摩尔浓度为1mol/l;
[0095]
2、将无水乙醇和对苯醌混合,搅拌至完全溶解,超声分散均匀后,制得0.3mol/l的对苯醌乙醇溶液;
[0096]
3、在磁力搅拌条件下,将对苯醌乙醇溶液逐滴滴加到第一混合溶液中,室温下搅拌30min后,在搅拌的第二混合溶液中滴加质量浓度为68%的浓硝酸,调节ph值为3.8,得到电化学沉积液;
[0097]
4、采用所述电化学沉积液进行电化学沉积;
[0098]
所述电化学沉积采用三电极体系,以ag/agcl为参比电极,pt片为对电极,fto为工作电极,以所述电化学沉积液为电解液;在沉积过程采用恒电位法;所述电化学沉积的沉积电位为-0.1v
ag/agcl
,沉积时间为300s;
[0099]
电化学沉积后,将所述薄膜前体依次用去离子水和乙醇冲洗,干燥后薄膜前体;
[0100]
5、将所述薄膜前体在250℃下退火处理1h,得到卤氧化铋固溶体光电薄膜biobr
xi1-x
(x=0.2),命名为s2。
[0101]
实施例3
[0102]
1、向硝酸铋中加水,搅拌至完全溶解,超声分散均匀后,制得20mmol/l的硝酸铋水溶液;量取100ml上述硝酸铋水溶液,并加入碘化钠和溴化钠(碘化钠和溴化钠的摩尔比为3:2),超声分散均匀,得到第一混合溶液;所述第一混合溶液中卤化钠(包括碘化钠和溴化钠)的摩尔浓度为1mol/l;
[0103]
2、将无水乙醇和对苯醌混合,搅拌至完全溶解,超声分散均匀后,制得0.3mol/l的对苯醌乙醇溶液;
[0104]
3、在磁力搅拌条件下,将对苯醌乙醇溶液逐滴滴加到第一混合溶液中,室温下搅拌30min后,在搅拌的第二混合溶液中滴加质量浓度为68%的浓硝酸,调节ph值为3.4,得到电化学沉积液;
[0105]
4、采用所述电化学沉积液进行电化学沉积;
[0106]
所述电化学沉积采用三电极体系,以ag/agcl为参比电极,pt片为对电极,fto为工作电极,以所述电化学沉积液为电解液;在沉积过程采用恒电位法;所述电化学沉积的沉积电位为-0.1v
ag/agcl
,沉积时间为300s;
[0107]
电化学沉积后,将所述薄膜前体依次用去离子水和乙醇冲洗,干燥后薄膜前体;
[0108]
5、将所述薄膜前体在250℃下退火处理1h,得到卤氧化铋固溶体光电薄膜biobr
xi1-x
(x=0.4),命名为s3。
[0109]
实施例4
[0110]
1、向硝酸铋中加水,搅拌至完全溶解,超声分散均匀后,制得20mmol/l的硝酸铋水溶液;量取100ml上述硝酸铋水溶液,并加入碘化钠和溴化钠(碘化钠和溴化钠的摩尔比为2:3),超声分散均匀,得到第一混合溶液;所述第一混合溶液中卤化钠(包括碘化钠和溴化钠)的摩尔浓度为1mol/l;
[0111]
2、将无水乙醇和对苯醌混合,搅拌至完全溶解,超声分散均匀后,制得0.3mol/l的对苯醌乙醇溶液;
[0112]
3、在磁力搅拌条件下,将对苯醌乙醇溶液逐滴滴加到第一混合溶液中,室温下搅拌30min后,在搅拌的第二混合溶液中滴加质量浓度为68%的浓硝酸,调节ph值为2.8,得到电化学沉积液;
[0113]
4、采用所述电化学沉积液进行电化学沉积;
[0114]
所述电化学沉积采用三电极体系,以ag/agcl为参比电极,pt片为对电极,fto为工作电极,以所述电化学沉积液为电解液;在沉积过程采用恒电位法;所述电化学沉积的沉积电位为-0.1v
ag/agcl
,沉积时间为300s;
[0115]
电化学沉积后,将所述薄膜前体依次用去离子水和乙醇冲洗,干燥后薄膜前体;
[0116]
5、将所述薄膜前体在250℃下退火处理1h,得到卤氧化铋固溶体光电薄膜
biobr
xi1-x
(x=0.6),命名为s4。
[0117]
实施例5
[0118]
1、向硝酸铋中加水,搅拌至完全溶解,超声分散均匀后,制得20mmol/l的硝酸铋水溶液;量取100ml上述硝酸铋水溶液,并加入碘化钠和溴化钠(碘化钠和溴化钠的摩尔比为1:4),超声分散均匀,得到第一混合溶液;所述第一混合溶液中卤化钠(包括碘化钠和溴化钠)的摩尔浓度为1mol/l;
[0119]
2、将无水乙醇和对苯醌混合,搅拌至完全溶解,超声分散均匀后,制得0.3mol/l的对苯醌乙醇溶液;
[0120]
3、在磁力搅拌条件下,将对苯醌乙醇溶液逐滴滴加到第一混合溶液中,室温下搅拌30min后,在搅拌的第二混合溶液中滴加质量浓度为68%的浓硝酸,调节ph值为2.4,得到电化学沉积液;
[0121]
4、采用所述电化学沉积液进行电化学沉积;
[0122]
所述电化学沉积采用三电极体系,以ag/agcl为参比电极,pt片为对电极,fto为工作电极,以所述电化学沉积液为电解液;在沉积过程采用恒电位法;所述电化学沉积的沉积电位为-0.1v
ag/agcl
,沉积时间为300s;
[0123]
电化学沉积后,将所述薄膜前体依次用去离子水和乙醇冲洗,干燥后薄膜前体;
[0124]
5、将所述薄膜前体在250℃下退火处理1h,得到卤氧化铋固溶体光电薄膜biobr
xi1-x
(x=0.8),命名为s5。
[0125]
实施例6
[0126]
1、向硝酸铋中加水,搅拌至完全溶解,超声分散均匀后,制得20mmol/l的硝酸铋水溶液;量取100ml上述硝酸铋水溶液,并加入溴化钠,超声分散均匀,得到第一混合溶液;所述第一混合溶液中溴化钠的摩尔浓度为1mol/l;
[0127]
2、将无水乙醇和对苯醌混合,搅拌至完全溶解,超声分散均匀后,制得0.3mol/l的对苯醌乙醇溶液;
[0128]
3、在磁力搅拌条件下,将对苯醌乙醇溶液逐滴滴加到第一混合溶液中,室温下搅拌30min后,在搅拌的第二混合溶液中滴加质量浓度为68%的浓硝酸,调节ph值为2.0,得到电化学沉积液;
[0129]
4、采用所述电化学沉积液进行电化学沉积;
[0130]
所述电化学沉积采用三电极体系,以ag/agcl为参比电极,pt片为对电极,fto为工作电极,以所述电化学沉积液为电解液;在沉积过程采用恒电位法;所述电化学沉积的沉积电位为-0.1v
ag/agcl
,沉积时间为300s;
[0131]
电化学沉积后,将所述薄膜前体依次用去离子水和乙醇冲洗,干燥后薄膜前体;
[0132]
5、将所述薄膜前体在250℃下退火处理1h,得到卤氧化铋固溶体光电薄膜biobr
xi1-x
(x=1),命名为s6。
[0133]
试验例
[0134]
1、分别对实施例1~6得到的样品s1~s6进行x射线衍射、eds能谱分析
[0135]
图1为本发明实施例中样品s1和样品s6的xrd图谱以及对应的标准pdf卡片。通过将s1的xrd图谱与bioi的标准pdf卡片比较可知,样品s1为纯相bioi,通过将s6的xrd图谱与biobr的标准pdf卡片比较可知,样品s6为纯相biobr。
[0136]
图2为本发明实施例中样品s1~s6的xrd图谱。通过将样品s1~s6的xrd图谱进行对比可知,样品s2~s5在22~35
°
的衍射峰处于样品s1和s6的(101)、(102)、(110)峰之间,且峰位随br:i比例的升高而逐渐向高角度偏移,这种峰位偏移是由于br-的离子半径(0.196nm)小于i-的离子半径(0.220nm),br-取代bioi晶格中的i-引起晶格常数变小,晶面衍射角度增大。
[0137]
图3为本发明实施例中样品s1~s6的eds元素分析对比图。从图3可知,所获得的样品的br:i比例可通过投料比进行调控,表明biobr
xi1-x
(0≤x≤1)固溶体光电薄膜的成功制备。
[0138]
2、对样品s1~s6的紫外可见光吸收图谱和光学带隙进行分析
[0139]
图4为本发明实施例中样品s1~s6的颜色对比照片。从图4可知,随着biobr
xi1-x
(0≤x≤1)固溶体光电薄膜中br:i比例的增加,样品颜色逐渐由橙红色变为灰白。
[0140]
图5为本发明实施例中样品s1~s6的紫外可见光吸收光谱图。从图5可知,随着biobr
xi1-x
(0≤x≤1)固溶体光电薄膜中br:i比例的增加,样品的光吸收带边逐渐由长波长波段向短波长波段移动,由637nm转变为437nm。
[0141]
图6为实施例中样品s1~s6的tauc图。从图6可知,随着biobr
xi1-x
(0≤x≤1)固溶体光电薄膜中br:i比例的增加,样品的光学带隙逐渐由1.84ev增加为2.6ev。
[0142]
3、对样品s1~s6的微观形貌进行扫描电镜分析
[0143]
图7为本发明实施例中样品s1~s6的sem图。从图7可知,随着biobr
xi1-x
(0≤x≤1)固溶体光电薄膜中br:i比例的增加,薄膜样品的微观形貌逐渐由bioi的密集直立式短片状片层形貌向biobr的疏松弯曲式薄片状片层形貌过渡。
[0144]
4、对样品s1~s6的能带结构进行分析
[0145]
图8为本发明实施例中样品s1~s6的莫特肖特基曲线图。从图8可知,所制备的biobr
xi1-x
(0≤x≤1)固溶体光电薄膜均为n型半导体,且随着biobr
xi1-x
(0≤x≤1)固溶体光电薄膜中br:i比例的增加,薄膜样品的平带电位相对于可逆氢电极电位处于-0.24~-0.48v之间。
[0146]
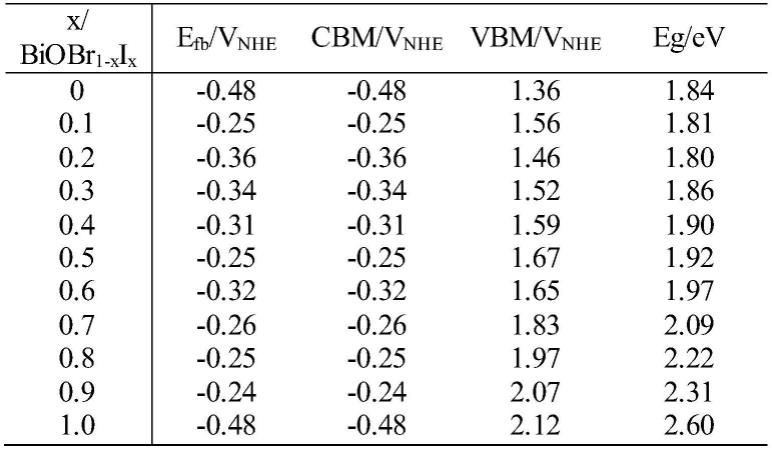
通过对薄膜样品的平带电位及光学带隙的表征,获得本发明实施例中样品s1~s6的能带结构表及能带结构示意图,分别如表1、图9所示。图9为本发明实施例中样品s1~s6的能带结构示意图。
[0147]
表1本发明实施例中样品s1~s6的能带结构表
[0148][0149]
从表1和图9可知,随着biobr
xi1-x
(0≤x≤1)固溶体光电薄膜中br:i比例的增加,薄膜样品的价带位置产生正向偏移。
[0150]
5、对样品s1的i-v曲线进行检测
[0151]
在三电极体系中,以ag/agcl为参比电极,pt片为对电极,样品s1为工作电极,以1mol/lna2so4和1mol/lna2so3的混合溶液为电解液,以am 1.5g太阳光模拟器为光源,对s1样品的i-v曲线进行检测,结果如图10所示。图10为本发明实施例中样品s1退火处理前后的i-v曲线图。退火后的s1样品,当外加偏压相对于ag/agcl参比电极为0v时,产生的光电流密度可达1.1ma/cm2;当外加偏压相对于ag/agcl参比电极为0.6v时,产生的光电流密度可达2ma/cm2。
[0152]
6、对实施例中样品s5的光催化、电催化及光电催化降解罗丹明b的降解性能进行测试
[0153]
以20ml浓度为5mg/l的阳离子染料罗丹明b(rhb)为测试溶液,其中电催化及光电催化降解需在测试溶液中加入0.1mol/l的na2so4,样品s5的面积为1
×
1cm2。光催化降解以300w的xe灯作为实验光源,每次测量前,反应体系需要在黑暗中保持30min,在连续搅拌下建立吸附和解吸之间的平衡,之后打开xe灯光源并每隔1小时取750μl反应液,在uv-vis分光光度计上利用550nm处的特征吸收峰分析残留rhb浓度。电催化降解采用三电极体系,ag/agcl为参比电极,铂网为对电极,样品s5光电极为工作电极,整个测量在黑暗中进行,测量前反应体系需要在黑暗中保持30min,在连续搅拌下建立吸附和解吸之间的平衡,之后通过电化学工作站为样品s5施加相对于ag/agcl参比电极为1.0v的偏压,然后每隔1小时取750μl反应液,在uv-vis分光光度计上利用550nm处的特征吸收峰分析残留rhb浓度。光电催化降解同样采用三电极体系,并以300w的xe灯作为实验光源,每次测量前,反应体系需要在黑暗中保持30min,在连续搅拌下建立吸附和解吸之间的平衡,之后打开xe灯并利用电化学工作站为样品s5施加相对于ag/agcl参比电极为1.0v的偏压,然后每隔1小时取750μl反应液,在uv-vis分光光度计上利用550nm处的特征吸收峰分析残留rhb浓度。图11为本发明实施例中样品s5的光催化、电催化及光电催化降解rhb的降解性能。样品s5在光催化(pc)和电催化(ec)过程中对rhb分别只降解了22.8%和11.7%,而光电催化(pec)的降解率为63.4%,远高于二者。图12为本发明实施例中样品s5的光催化、电催化及光电催化降解rhb的降解动力
学曲线。s5样品的pec降解速率阐述分别是pc(0.09h-1
)的3.5倍、ec(0.042h-1
)的8.1倍。
[0154]
以上实施例的说明只是用于帮助理解本发明的方法及其核心思想。对这些实施例的多种修改对本领域的专业技术人员来说将是显而易见的,本文中所定义的一般原理可以在不脱离本发明的精神或范围的情况下,在其它实施例中实现。因此,本发明将不会被限制于本文所示的这些实施例,而是要符合与本文所公开的原理和新颖特点相一致的最宽的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1