一种从黄芪中分离制备毛蕊异黄酮的方法
1.本发明属于化合物提取纯化技术领域,具体涉及一种从黄芪中分离制备高纯度毛蕊异黄酮的方法。
背景技术:
2.黄芪作为传统补益药,具有补气升阳、益胃固表、利水消肿、脱毒生肌等功效。毛蕊异黄酮(calycosin,cys)是典型的异黄酮类化合物,是黄芪的主要活性成分之一,具有抗氧化、抗疟原虫、保护血管内皮细胞、抗高血压、抗高血糖、神经保护、增强造血功能、及治疗白血病等多方面药理作用,是鉴定黄芪药材及黄芪中药必不可少的的质量指标之一。然而,由于黄芪原料中毛蕊异黄酮含量较低,且存在多种黄酮类组分,从而使得制备高纯度的毛蕊异黄酮难度较大。截止目前,国内尚未有研究机构能提供毛蕊异黄酮标准样品及其制备方法,因此为毛蕊异黄酮含量测定制备相应标准样品尤为重要,对以毛蕊异黄酮为评价指标的药材、制剂质量评价和cys发挥药效防治相关疾病具有重要意义。
3.传统的分离制备毛蕊异黄酮的技术方法主要有硅胶柱层析法、高速逆流色谱法等。公开号为cn101775418 a采用匀浆萃取-混合酶诱导生物转化技术、负压空化提取技术、液液萃取、大孔吸附树脂、硅胶层析等技术,得到纯度为95%的毛蕊异黄酮对照品;cn102079738a精简步骤,通过反复硅胶层析结合重结晶技术,从黄芪提取物中得到纯度为98%的对照品,但上述方法存在操作繁琐,样品回收率低等问题。
4.高速逆流色谱作为一种连续高效的液-液分配色谱,具有无不可逆吸附,回收率高、进样量大等特点,已成为分离天然产物的极佳候选方法,可用于毛蕊异黄酮等黄酮类天然产物的分离纯化。
技术实现要素:
5.本发明的目的是为了提供一种从黄芪中分离制备毛蕊异黄酮的方法,即一种高纯度毛蕊异黄酮的制备及其质量控制方法,从而弥补现有技术的不足。
6.本发明所提供的高纯度毛蕊异黄酮的制备方法,包括以下步骤:
7.1)取黄芪粉末,加入乙醇溶液中进行超声提取,提取液减压蒸馏回收溶剂得到浸膏;
8.所述的乙醇溶液,作为实施例的一种具体记载,为70%乙醇溶液;
9.2)将步骤1)中制备的浸膏采用高速逆流色谱进行纯化,采用氯仿、甲醇、醋酸和水的溶液作为两相溶剂系统。按色谱峰收集目标组分,浓缩后冷冻干燥得到样品;
10.其中两相溶剂系统中氯仿、甲醇、醋酸和水的体积比为2:1:1:1~1.5。
11.上述高速逆流色谱法中溶剂系统上相为固定相,下相为流动相,高速逆流色谱分离条件如下:温度35℃,检测波长254nm,转速1000rpm,流动相采用流速梯度;
12.3)将步骤2)的样品以0.5-2.5ml的进样量上制备高效液相色谱,制备液相色谱条件如下:色谱柱为c18柱,以有机溶剂乙腈的水溶液作为洗脱剂洗脱,按色谱峰收集目标组
分,将流出液浓缩后冷冻干燥,得到毛蕊异黄酮;
13.上述制备型液相色谱5ml/min-8ml/min的流速下,流动相(体积比)乙腈:水=70:30或甲醇:水=50:50,检测波长为254nm。
14.本发明方法制备的毛蕊异黄酮纯度可达到99.90%以上,纯度符合化学对照品要求,为黄芪药材的质量控制提供科学基础和保证。本发明采用高速逆流色谱结合制备型高效液相色谱制备高纯度毛蕊异黄酮,分离速度快,生产周期短,可稳定获得纯度为99.90%以上毛蕊异黄酮,制备量大,适合工业化生产,有较好的应用前景。
附图说明
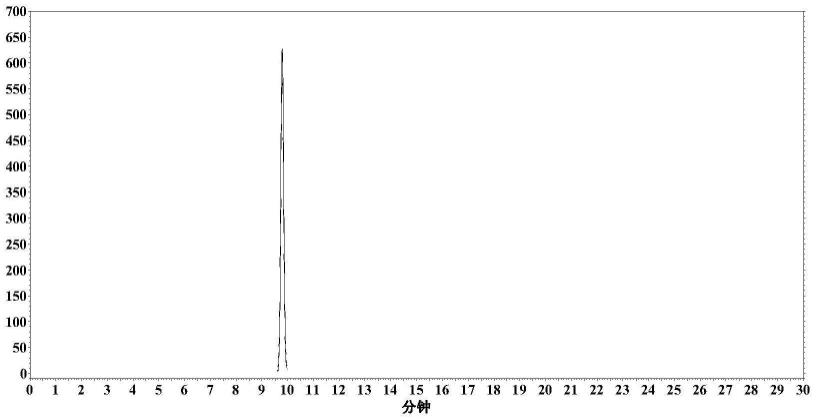
15.图1:高速逆流色谱纯化后毛蕊异黄酮的hplc图谱
16.图2:高效液相色谱纯化后毛蕊异黄酮的hplc图谱
具体实施方式
17.本发明的方法从黄芪中分离纯化高纯度毛蕊异黄酮,具有制备量大,周期短,纯度高等优点,具备可控性和自动化生产性,具有广泛的应用前景。
18.下面结合实施例对本发明进行详细的描述。
19.实施例1
20.取黄芪粉末1kg,粉碎,加入70%乙醇超声提取(料液比1:10,40khz,40℃)两次,每次1小时,合并提取液,减压蒸馏回收溶剂得浸膏。
21.所得浸膏进而采用高速逆流色谱进行分离纯化,以两相溶剂系统:氯仿:甲醇:乙酸:水(2:1:1:1,v/v/v/v)作为流动相;将上述比例溶剂在分液漏斗中剧烈振摇5min使其充分混合,然后静置过夜使其两相达到分配平衡后即得两相溶剂,其中上相为固定相,下相为流动相。首先设定流速为15ml
·
min-1
将固定相泵入hsccc主机的聚四氟乙烯色谱柱中,直至固定相从仪器尾端流出,停止泵液;设定仪器转速为1000rpm,待转速稳定后设定流速为1ml
·
min-1
开始泵入下相;待流动相从色谱柱尾端流出,表明柱内两相溶剂已达到流体动力学动态平衡;将样品溶液通过进样阀注入色谱仪进行分离。流动相流速采用梯度流速,前300min流速为1.0ml
·
min-1
,300min时切换为2.0ml
·
min-1
;转速为1000rpm;分离温度为35℃;进样量为500mg(25mg
·
ml-1
),检测波长为254nm,得毛蕊异黄酮样品,纯度为96.43%,如图1所示。
22.所得样品用制备型高效液相色谱进行制备,色谱柱采用c18柱(10mm
×
250mm,5μm),流动相:乙腈:水体积比为30:70,检测波长为254nm,流速为8ml/min,按色谱峰收集目标化合物,减压浓缩,收集毛蕊异黄酮白色粉末。
23.采用分析型高效液相色谱对收集得到的毛蕊异黄酮纯度进行检测,色谱柱:sinochrom ods-bp:流动相:甲醇:0.1%甲酸水体积比为50:50,检测波长为254nm,流速为1ml/min。测得产品纯度为99.93%,如图2所示。
24.实施例2:
25.取黄芪粉末2kg,粉碎,加入70%乙醇超声提取(料液比1:10,40khz,40℃)两次,每次3小时,合并提取液,减压蒸馏回收溶剂得浸膏。所得浸膏进而采用高速逆流色谱进行分离纯化,以两相溶剂系统:氯仿:甲醇:乙酸:水(2:1:1:1,v/v/v/v)作为流动相;将上述比例
溶剂在分液漏斗中剧烈振摇5min使其充分混合,然后静置过夜使其两相达到分配平衡后即得两相溶剂,其中上相为固定相,下相为流动相。首先设定流速为15ml
·
min-1
将固定相泵入hsccc主机的聚四氟乙烯色谱柱中,直至固定相从仪器尾端流出,停止泵液;设定仪器转速为1000rpm,待转速稳定后设定流速为1ml
·
min-1
开始泵入下相;待流动相从色谱柱尾端流出,表明柱内两相溶剂已达到流体动力学动态平衡;将样品溶液通过进样阀注入色谱仪进行分离。流动相流速采用梯度流速,前300min流速为1.0ml
·
min-1
,300min时切换为2.0ml
·
min-1
;转速为1000rpm;分离温度为35℃;进样量为500mg(25mg
·
ml-1
),检测波长为254nm,得毛蕊异黄酮样品,纯度为90.22%。所得样品用制备型高效液相色谱进行制备,色谱柱采用c18柱(10mm
×
250mm,5μm),流动相:乙腈:水体积比为30:70,检测波长为254nm,流速为8ml/min,按色谱峰收集目标化合物,减压浓缩,收集毛蕊异黄酮白色粉末。
26.采用分析型高效液相色谱对收集得到的毛蕊异黄酮纯度进行检测,色谱柱:sinochrom ods-bp:流动相:水体积比为50:50,检测波长为254nm,流速为1ml/min。测得产品纯度为99.90%。
27.实施例3
28.取黄芪粉末1kg,粉碎,加入70%乙醇超声提取(料液比1:10,40khz,40℃)两次,每次1小时,合并提取液,减压蒸馏回收溶剂得浸膏。所得浸膏进而采用高速逆流色谱进行分离纯化,以两相溶剂系统:氯仿:甲醇:乙酸:水(2:1:1:1.5,v/v/v/v)作为流动相;将上述比例溶剂在分液漏斗中剧烈振摇5min使其充分混合,然后静置过夜使其两相达到分配平衡后即得两相溶剂,其中上相为固定相,下相为流动相。首先设定流速为15ml
·
min-1
将固定相泵入hsccc主机的聚四氟乙烯色谱柱中,直至固定相从仪器尾端流出,停止泵液;设定仪器转速为1000rpm,待转速稳定后设定流速为1ml
·
min-1
开始泵入下相;待流动相从色谱柱尾端流出,表明柱内两相溶剂已达到流体动力学动态平衡;将样品溶液通过进样阀注入色谱仪进行分离。流动相流速采用梯度流速,前300min流速为1.0ml
·
min-1
,300min时切换为2.0ml
·
min-1
;转速为1000rpm;分离温度为35℃;进样量为500mg(25mg
·
ml-1
),检测波长为254nm,得毛蕊异黄酮样品,纯度为92.33%。所得样品用制备型高效液相色谱进行制备,色谱柱采用c18柱(10mm
×
250mm,5μm),流动相:甲醇:水体积比为50:50,检测波长为254nm,流速为8ml/min,按色谱峰收集目标化合物,减压浓缩,收集毛蕊异黄酮白色粉末。
29.采用分析型高效液相色谱对收集得到的毛蕊异黄酮纯度进行检测,色谱柱:sinochrom ods-bp:流动相:甲醇:0.1%甲酸水体积比为50:50,检测波长为254nm,流速为1ml/min。测得产品纯度为99.89%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1