一种哌嗪基碳捕集剂的合成方法
胺烷基哌嗪(化合物a)与脂肪酮(化合物b)的摩尔比为1:0.5~5,持续反应2h得到含有伯胺基保护的胺烷基哌嗪中间体的反应溶液,具体反应方程式如下所示:
[0012][0013]
所述搅拌方式、滴加方式和反应容器不做具体限制,为本领域技术人员熟知的方式即可。
[0014]
优选的,所述n-胺烷基哌嗪与脂肪酮的摩尔比为1:1~1.5。
[0015]
(2)加成反应:在30~80℃搅拌条件下,先向反应溶液中加入极性溶剂搅拌均匀,后向含有胺烷基哌嗪中间体的反应溶液中加入环氧烷烃(化合物c)进行亲核加成反应,控制环氧烷烃与n-胺烷基哌嗪的摩尔比为1~1.5:1,持续反应5h,得到含有伯胺基保护的羟烷基哌嗪中间体的反应溶液,具体反应方程式如下所示:
[0016][0017]
所述极性溶剂为甲醇、乙醇、丙醇、二氧六环和四氢呋喃中的一种。
[0018]
优选的,所述反应温度为30~50℃。
[0019]
所述加热方式、加料方式和反应容器不做具体限制,为本领域技术人员熟知的方式即可。
[0020]
(3)水解反应:在室温搅拌下,向含有羟烷基哌嗪中间体的反应溶液中加入蒸馏水,控制蒸馏水与n-胺烷基哌嗪的摩尔比为1:1~2:1,持续反应2h,得到含有羟烷基胺烷基哌嗪(化合物d)的反应溶液,具体反应方程式如下所示:
[0021][0022]
(4)产物的分离精制:将含有化合物d的反应溶液进行精馏操作,收集80~120℃馏分并在室温下冷却、静置分相,上相作脂肪酮回用,下相作蒸馏水回用;精馏残余液为目标化合物的粗产物,使用有机溶剂在20~50℃搅拌条件下溶解过量的粗产物,后在-10~0℃下进行重结晶操作,过滤,得到化合物d;
[0023]
所述重结晶操作所用有机溶剂为甲醇、乙醇、正丙醇、异丙醇、乙醚和正己烷中的一种或一种以上;优选为乙醇、乙醚和正己烷中的一种或一种以上;
[0024]
所述精馏装置、液-液分相装置及重结晶装置不做具体限制,为本领域技术人员熟知的方式即可。
[0025]
所述哌嗪基碳捕集剂的制备操作为:常温搅拌下,按照质量分数为5~70%将化合物d加入水中,与添加剂、助剂复配,即制得哌嗪基碳捕集剂。
[0026]
以本发明合成的哌嗪基有机胺碳捕集剂或以本发明所述合成方法合成的羟烷基胺烷基哌嗪为主要成分的复合型碳捕集剂用于燃煤发电、钢铁、冶金、水泥建材、石油化工、玻璃、超细粉体等产业工业烟气中二氧化碳的捕集、回收与利用。
[0027]
与现有技术相比,本发明的有益效果是:
[0028]
1.本发明所述的单端基保护合成方法平均产率高于85%,产物纯度高于98%。
[0029]
2.本发明所述的胺基保护剂价廉易得,可循环利用,具有一定的经济效益。
附图说明
[0030]
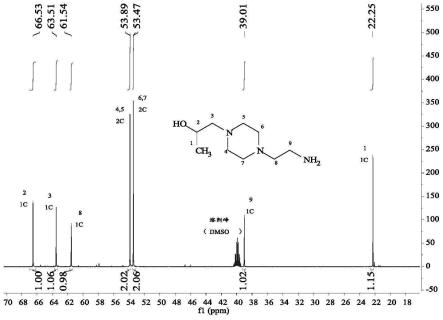
图1为本发明中1-(2-羟丙基)-4-(2-胺乙基)哌嗪的
13
b nmr谱图;
[0031]
图2为本发明中1-(2-羟丙基)-4-(2-胺乙基)哌嗪的1h nmr谱图;
[0032]
图3为本发明中提纯后的1-(2-羟丙基)-4-(2-胺乙基)哌嗪气相色谱图。
具体实施方式
[0033]
下面将结合本发明实施例对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0034]
实施例1
[0035]
当化合物d中r1=亚乙基,r2=甲基(具体化合物为1-(2-羟丙基)-4-(2-胺乙基)哌嗪);化合物b中r3=甲基,r4=异丁基(具体化合物为甲基异丁基甲酮)时,其合成原理为:
[0036][0037]
具体操作步骤为:向500ml三口烧瓶内加入0.1mol的n-胺乙基哌嗪和1.5mol的甲基异丁基甲酮,控制磁力搅拌转速为100r/min,待溶液混合均匀后,加蒸馏装置并加热溶液至110℃,先收集87~90℃馏分至无液体蒸出后,再收集105~108℃馏分至无液体蒸出,将馏出液收集于锥形瓶中等待回收处理。向剩余的反应溶液中加入2mol甲醇,待溶液搅拌均匀后,控制温度为30℃,用恒压漏斗缓慢向三口烧瓶中滴加0.15mol环氧丙烷,1h内加完,持续反应3h后,加蒸馏装置,控制溶液温度为80℃,将甲醇和未参与反应的环氧丙烷蒸出。后向反应溶液中加入1mol蒸馏水,控制磁力搅拌转速为500r/min,反应2h后,后控制溶液温度为110℃,收集105~108℃馏分直至再无馏分蒸出,将馏出液收集于锥形瓶中等待回收处理。将蒸馏残余液冷却至室温,向三口烧瓶中加入0.1mol乙醇,待溶液混合均匀后,控制温度为-10℃,静置2h有大量淡黄色晶体析出,过滤,得到1-(2-羟丙基)-4-(2-胺乙基)哌嗪。产物总收率为92%,纯度为98.3%。产物核磁表征结果如图1和图2所示,气相色谱表征结果
如图3所示。
[0038]
将上述蒸馏操作中得到的待处理的甲基异丁基甲酮和水的混合溶液在500ml分液漏斗中混匀后静置2h,得到澄清的上、下两相混合溶液,下相从底部流出,此下相为水相,后作脱保护溶剂回用;上相从分液漏斗顶部倒出,此上相为有机相,后作胺基保护剂回用。气相色谱检测结果:上相甲基异丁基甲酮质量分数95.7%,下相甲基异丁基甲酮质量分数0.81%。
[0039]
实施例2
[0040]
当化合物d中r1=亚乙基,r2=h质子(具体化合物为1-(2-羟乙基)-4-(2-胺乙基)哌嗪);化合物b中r3=甲基,r4=异丁基(具体化合物为甲基异丁基甲酮)时,其合成原理为:
[0041][0042]
向500ml三口烧瓶内加入0.1mol的n-胺乙基哌嗪和150g经回收的胺基保护剂(甲基异丁基甲酮质量分数为95.7%),控制磁力搅拌转速为100r/min,待溶液混合均匀后,加蒸馏装置并加热溶液至110℃,先收集87~90℃馏分至无液体蒸出后,再收集105~108℃馏分至无液体蒸出,将馏出液收集于锥形瓶中等待回收处理。向剩余的反应溶液中加入2mol甲醇,待溶液搅拌均匀后,转移至高压反应釜内,控制釜内温度为25℃,控制气体流量计为1ml/min,向釜内累计通入0.1mol环氧乙烷气体。控制磁力搅拌转速为100r/min,持续反应3h,反应完毕。将釜内反应溶液转移至250ml三口烧瓶中,加蒸馏装置,控制溶液温度为80℃,将甲醇蒸出。后向反应溶液中加入18g经回收的脱保护溶剂(水的质量分数为98.8%),控制磁力搅拌转速为500r/min,反应2h后,加蒸馏装置,控制溶液温度为110℃,收集105~108℃馏分直至再无馏分蒸出,将馏出液收集于锥形瓶中等待回收处理。将蒸馏残余液冷却至室温后,向其中加入0.1mol乙醇,待溶液混合均匀后,控制温度为-10℃,静置2h有大量白色晶体析出,过滤,得到1-(2-羟乙基)-4-(2-胺乙基)哌嗪。产物总收率为86%,纯度为97.6%。
[0043]
本实施例中保护剂和脱保护溶剂的回收利用步骤与实施例1中的步骤一致,气相色谱检测结果:上相甲基异丁基甲酮质量分数96.8%,下相甲基异丁基甲酮质量分数1.03%。
[0044]
实施例3
[0045]
当化合物d中r1=亚甲基,r2=甲基(具体化合物为1-(2-羟丙基)-4-胺甲基哌嗪);化合物b中r3=甲基,r4=异丁基(具体化合物为甲基异丁基甲酮)时,其合成、分离和精制纯化步骤与实施例1基本一致,不同点为将作为原料的n-胺乙基哌嗪替换为n-胺甲基哌嗪。最终产物总收率为90%,纯度为99.3%。
[0046]
保护剂的回收利用步骤与实施例1中的步骤一致,气相色谱检测结果:上相甲基异
丁基甲酮质量分数98.8%,下相甲基异丁基甲酮质量分数0.87%。
[0047]
实施例4
[0048]
当化合物d中r1=亚甲基,r2=h质子(具体化合物为1-(2-羟乙基)-4-胺甲基哌嗪);化合物b中r3=甲基,r4=异丁基(具体化合物为甲基异丁基甲酮)时,其合成、分离和精制纯化步骤与实施例2基本一致,不同点为将作为原料的n-胺乙基哌嗪替换为n-胺甲基哌嗪。最终产物总收率为84%,纯度为98.9%。
[0049]
保护剂的回收利用步骤与实施例1中的步骤一致,气相色谱检测结果:上相甲基异丁基甲酮质量分数97.2%,下相甲基异丁基甲酮质量分数0.94%。
[0050]
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,根据本发明的技术方案及其发明构思加以等同替换或改变,都应涵盖在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1