一种使用聚合物/二氧化硅负载的胺中空纤维吸附剂制备方法与流程
1.本发明涉及二氧化碳吸附净化领域,具体是一种使用聚合物/二氧化硅负载的胺中空纤维吸附剂制备方法。
背景技术:
2.目前,已有各种各样的捕获材料应用到二氧化碳捕获过程中,主要包括二氧化碳吸收溶剂、二氧化碳分离膜、二氧化碳固体吸附剂等。
3.二氧化碳吸收剂是所有分离捕集二氧化碳材料中出现最早、技术最成熟、应用最广泛的材料。按照溶剂吸收过程的原理不同,二氧化碳吸收溶剂包括物理吸收剂和化学吸收剂。利用溶剂吸收二氧化碳的吸收过程是通过吸收溶液对混合气体中的二氧化碳进行选择性的吸收或者洗脱,最终将二氧化碳从混合气体中脱除或浓缩的过程。
4.用于二氧化碳的分离膜要求不仅具有好的透过率,而且二氧化碳在膜上的选择性效果要好。因为烟气中的主要成分是氮气和二氧化碳,两种气体的分子大小相差很小,所以不容易从中捕集二氧化碳。因此,膜分离过程中首要注意的问题是高渗透通量和高选择性的同时获得。
5.利用固体材料对混合气体中c02进行的选择性吸附,然后在不同的再生条件下将c02解吸出来,从而使固体材料得到可再生循环利用。其中的固体物质成吸附剂,被吸附的物质称为吸附质。相比于其他二氧化碳捕集材料,二氧化碳固体吸附剂来源广泛,工艺过程简单,能耗低,无设备腐蚀和环境污染,压力适应范围广;常温下即可操作,省去了加热和冷却的能量耗损,而且灵活性强,操作弹性大;吸附剂可循环再生性强。因此,二氧化碳固体吸附剂得到了广泛的研究与利用。
6.本发明抛弃传统的二氧化硅微球,将二氧化硅制成多孔、中空的二氧化硅纳米纤维,在将聚合物接枝在二氧化硅纳米纤维,再进行胺基的负载。本发明制备的吸附剂虽然聚合物的接枝使二氧化硅纳米纤维的孔隙率变小,但胺基的加入使得二氧化碳的吸附性能大大增加。本发明对高效的二氧化碳吸附剂的制备具有重要的意义。
技术实现要素:
7.针对现有技术的不足,本发明的目的是提供一种使用聚合物/二氧化硅负载的胺中空纤维吸附剂制备方法,制备的吸附剂具有优异的比表面积、良好的孔隙率、高的吸附容量。
8.本发明的第一个方面提供了一种中空、多孔的二氧化硅纳米纤维的制备方法,其方法包括如下步骤:
9.将无水乙醇,正硅酸乙酯及聚环氧乙烷加于三口瓶中,30-50℃下,在恒温磁力水浴锅中搅拌,至溶液均匀透明后,向反应釜中滴加去离子水,溶液立即变浑独,即发生溶胶凝胶反应,继续搅拌,3-5h后溶液呈半透明状态,取出静置,溶液分层,上层为乳白半透明溶
液,下层析出物浓度较大呈微黄色;将下层微黄色析出物即聚环氧乙烷/二氧化硅混合液装入10ml的医用注射器中,医用注射器控制流速为0.3-0.7mm/s,进行电纺,纺丝条件为电压为8-16kv,接收距离为15-25cm,电纺得到聚乙烯醇/埃洛石纳米管改性复合纤维;取出纤维膜至铁盘中盛放,置于马弗炉内胆中,设定好升温程序后开始灼烧去除,升温程序为:以1.5℃/min的升温速率到200℃,保温2h,再以3.5℃/min的速率升高到600℃保温3-5h,随炉冷却。
10.在进行静电纺丝的过程中,优选的:医用注射器控制流速为0.5mm/s,电纺时的电压为12kv,接受距离为20cm。
11.在进行煅烧的过程中,优选的:以3.5℃/min的速率升高到600℃保温4h。
12.本发明的第二个方面提供了一种使用聚合物/二氧化硅负载的胺中空纤维吸附剂制备方法。制备方法如下:
13.称取一定量的二氧化硅纳米纤维膜,用100ml的n,n-二甲基甲酰胺溶解后倒入500ml三口烧瓶中超声分散30min,称取聚丙烯酸,用100ml的n,n-二甲基甲酰胺溶解后倒入三口烧瓶与二氧化硅纳米纤维混合,将混合溶液再超声分散30min后置于油浴中,将0.1-1g的4-二甲氨基吡啶和1-5g的n,n'-二环己基碳二亚胺分别用10ml的n,n-二甲基甲酰胺溶解后逐滴加入到反应体系中,在机械搅拌下反应12-15h,将反应物抽滤,用无水乙醇洗涤3次,置于真空烘箱中60℃烘干,得到二氧化硅-聚丙烯酸中间体;胺基改性二氧化硅-聚丙烯酸中间体:称取一定量的胺,用100ml的n,n-二甲基甲酰胺将其溶解于500ml三口烧瓶中,置于冰浴中,在强搅拌下将二氧化硅-聚丙烯酸中间体的n,n-二甲基甲酰胺溶液逐滴加入到反应体系中,滴加完毕后继续反应24小时,将反应后的产物用抽滤瓶抽滤出来,用无水乙醇洗涤遍,最后置于60℃真空烘箱中干燥后得到最终产物。
14.其中优选的:油浴温度控制在120-200℃;更为优选的:油浴温度控制在120℃。
15.优选的:二氧化硅-聚丙烯酸中间体的n,n-二甲基甲酰胺溶液的滴加速度为60-80滴/min;更为优选的:二氧化硅-聚丙烯酸中间体的n,n-二甲基甲酰胺溶液的滴加速度为60滴/min。
16.与现有技术相比,本发明具有以下有益效果:
17.1.本发明制备的吸附剂为一种中空、多孔的二氧化硅纳米纤维,其相较于普通的二氧化硅吸附剂具有较大的比表面积和孔隙率。
18.2.本发明制备的吸附剂,制备流程简单,生产成本较低,有利于批量生产。
19.3.本发明制备的吸附剂吸附容量高、吸附选择性好。
20.4.本发明制备的吸附剂操作简单,吸附可循环性强,压力适应范围广。
21.5.本发明制备的吸附剂具有足够的机械强度,化学稳定性及热稳定性。
附图说明
22.图1为本发明制备的实施例1的纳米纤维扫描图。
23.图2为本发明制备的实施例2的纳米纤维扫描图。
24.图3为本发明制备的实施例3的纳米纤维扫描图。
25.图4为本发明制备的对比例1(a)和实施例1(b)在40℃条件下的二氧化碳吸附曲线。
26.图5为本发明制备的实施例1和对比例1和2制得的二氧化碳吸附曲线。
27.图6为本发明制备的实施例1进行吸附循环测试制得的吸附容量图。
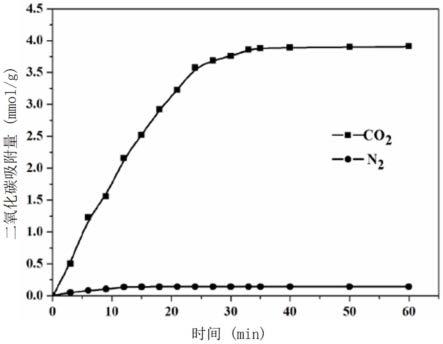
28.图7为本发明制备的实施例1对二氧化碳和氮气的吸附平衡曲线。
具体实施方式
29.以下通过特定的具体实例说明本发明的实施方式,本领域技术人员可由本说明书所揭露的内容轻易地了解本发明的其他优点与功效。本发明还可以通过另外不同的具体实施方式加以实施或应用,本说明书中的各项细节也可以基于不同观点与应用,在没有背离本发明的精神下进行各种修饰或改变。需说明的是,在不冲突的情况下,以下实施例及实施例中的特征可以相互组合。
30.实施例1
31.将40ml无水乙醇,4.46ml正硅酸乙酯及2g聚环氧乙烷加于三口瓶中,30-50℃下,在恒温磁力水浴锅中搅拌,至溶液均匀透明后,向反应釜中滴加去离子水,溶液立即变浑独,即发生溶胶凝胶反应,继续搅拌,5h后溶液呈半透明状态,取出静置,溶液分层,上层为乳白半透明溶液,下层析出物浓度较大呈微黄色;将下层微黄色析出物即聚环氧乙烷/二氧化硅混合液装入10ml的医用注射器中,医用注射器控制流速为0.5mm/s,进行电纺,纺丝条件为电压为12kv,接收距离为20cm,电纺得到聚乙烯醇/埃洛石纳米管改性复合纤维;取出纤维膜至铁盘中盛放,置于马弗炉内胆中,设定好升温程序后开始灼烧去除,升温程序为:以1.5℃/min的升温速率到200℃,保温2h,再以3.5℃/min的速率升高到600℃保温4h,随炉冷却。
32.称取3g二氧化硅纳米纤维膜,用100ml的n,n-二甲基甲酰胺溶解后倒入500ml三口烧瓶中超声分散30min,称取聚丙烯酸,用100ml的n,n-二甲基甲酰胺溶解后倒入三口烧瓶与二氧化硅纳米纤维混合,将混合溶液再超声分散30min后置于油浴中,将0.1g的4-二甲氨基吡啶和1g的n,n-二环己基碳二亚胺分别用10ml的n,n-二甲基甲酰胺溶解后逐滴加入到反应体系中,在机械搅拌下反应12h,将反应物抽滤,用无水乙醇洗涤3次,置于真空烘箱中60℃烘干,得到二氧化硅-聚丙烯酸中间体;胺基改性二氧化硅-聚丙烯酸中间体:10g的二乙醇胺,用100ml的n,n-二甲基甲酰胺将其溶解于500ml三口烧瓶中,置于冰浴中,在强搅拌下将二氧化硅-聚丙烯酸中间体的n,n-二甲基甲酰胺溶液逐滴加入到反应体系中,滴加完毕后继续反应24小时,将反应后的产物用抽滤瓶抽滤出来,用无水乙醇洗涤遍,最后置于60℃真空烘箱中干燥后得到最终产物。
33.实施例2
34.将40ml无水乙醇,4.46ml正硅酸乙酯及2g聚环氧乙烷加于三口瓶中,50℃下,在恒温磁力水浴锅中搅拌,至溶液均匀透明后,向反应釜中滴加去离子水,溶液立即变浑独,即发生溶胶凝胶反应,继续搅拌,3h后溶液呈半透明状态,取出静置,溶液分层,上层为乳白半透明溶液,下层析出物浓度较大呈微黄色;将下层微黄色析出物即聚环氧乙烷/二氧化硅混合液装入10ml的医用注射器中,医用注射器控制流速为0.3mm/s,进行电纺,纺丝条件为电压为15kv,接收距离为25cm,电纺得到聚乙烯醇/埃洛石纳米管改性复合纤维;取出纤维膜至铁盘中盛放,置于马弗炉内胆中,设定好升温程序后开始灼烧去除,升温程序为:以1.5℃/min的升温速率到200℃,保温2h,再以3.5℃/min的速率升高到600℃保温4h,随炉冷却。
35.称取3g二氧化硅纳米纤维膜,用100ml的n,n-二甲基甲酰胺溶解后倒入500ml三口烧瓶中超声分散30min,称取聚丙烯酸,用100ml的n,n-二甲基甲酰胺溶解后倒入三口烧瓶与二氧化硅纳米纤维混合,将混合溶液再超声分散30min后置于油浴中,将0.1g的4-二甲氨基吡啶和1g的n,n'-二环己基碳二亚胺分别用10ml的n,n-二甲基甲酰胺溶解后逐滴加入到反应体系中,在机械搅拌下反应12h,将反应物抽滤,用无水乙醇洗涤3次,置于真空烘箱中60℃烘干,得到二氧化硅-聚丙烯酸中间体;胺基改性二氧化硅-聚丙烯酸中间体:称取10g的二乙醇胺,用100ml的n,n-二甲基甲酰胺将其溶解于500ml三口烧瓶中,置于冰浴中,在强搅拌下将二氧化硅-聚丙烯酸中间体的n,n-二甲基甲酰胺溶液逐滴加入到反应体系中,滴加完毕后继续反应24小时,将反应后的产物用抽滤瓶抽滤出来,用无水乙醇洗涤遍,最后置于60℃真空烘箱中干燥后得到最终产物。
36.实施例3
37.将40ml无水乙醇,4.46ml正硅酸乙酯及2g聚环氧乙烷加于三口瓶中,30℃下,在恒温磁力水浴锅中搅拌,至溶液均匀透明后,向反应釜中滴加去离子水,溶液立即变浑独,即发生溶胶凝胶反应,继续搅拌,3h后溶液呈半透明状态,取出静置,溶液分层,上层为乳白半透明溶液,下层析出物浓度较大呈微黄色;将下层微黄色析出物即聚环氧乙烷/二氧化硅混合液装入10ml的医用注射器中,医用注射器控制流速为0.7mm/s,进行电纺,纺丝条件为电压为8kv,接收距离为15cm,电纺得到聚乙烯醇/埃洛石纳米管改性复合纤维;取出纤维膜至铁盘中盛放,置于马弗炉内胆中,设定好升温程序后开始灼烧去除,升温程序为:以1.5℃/min的升温速率到200℃,保温2h,再以3.5℃/min的速率升高到600℃保温4h,随炉冷却。
38.称取3g二氧化硅纳米纤维膜,用100ml的n,n-二甲基甲酰胺溶解后倒入500ml三口烧瓶中超声分散30min,称取聚丙烯酸,用100ml的n,n-二甲基甲酰胺溶解后倒入三口烧瓶与二氧化硅纳米纤维混合,将混合溶液再超声分散30min后置于油浴中,将0.1g的4-二甲氨基吡啶和1g的n,n'-二环己基碳二亚胺分别用10ml的n,n-二甲基甲酰胺溶解后逐滴加入到反应体系中,在机械搅拌下反应12h,将反应物抽滤,用无水乙醇洗涤3次,置于真空烘箱中60℃烘干,得到二氧化硅-聚丙烯酸中间体;胺基改性二氧化硅-聚丙烯酸中间体:称取10g的二乙醇胺,用100ml的n,n-二甲基甲酰胺将其溶解于500ml三口烧瓶中,置于冰浴中,在强搅拌下将二氧化硅-聚丙烯酸中间体的n,n-二甲基甲酰胺溶液逐滴加入到反应体系中,滴加完毕后继续反应24小时,将反应后的产物用抽滤瓶抽滤出来,用无水乙醇洗涤遍,最后置于60℃真空烘箱中干燥后得到最终产物。
39.实施例4
40.将40ml无水乙醇,4.46ml正硅酸乙酯及2g聚环氧乙烷加于三口瓶中,30℃下,在恒温磁力水浴锅中搅拌,至溶液均匀透明后,向反应釜中滴加去离子水,溶液立即变浑独,即发生溶胶凝胶反应,继续搅拌,3h后溶液呈半透明状态,取出静置,溶液分层,上层为乳白半透明溶液,下层析出物浓度较大呈微黄色;将下层微黄色析出物即聚环氧乙烷/二氧化硅混合液装入10ml的医用注射器中,医用注射器控制流速为0.5mm/s,进行电纺,纺丝条件为电压为12kv,接收距离为20cm,电纺得到聚乙烯醇/埃洛石纳米管改性复合纤维;取出纤维膜至铁盘中盛放,置于马弗炉内胆中,设定好升温程序后开始灼烧去除,升温程序为:以1.5℃/min的升温速率到200℃,保温2h,再以3.5℃/min的速率升高到600℃保温4h,随炉冷却。
41.称取3g二氧化硅纳米纤维膜,用100ml的n,n-二甲基甲酰胺溶解后倒入500ml三口
烧瓶中超声分散30min,称取聚丙烯酸,用100ml的n,n-二甲基甲酰胺溶解后倒入三口烧瓶与二氧化硅纳米纤维混合,将混合溶液再超声分散30min后置于油浴中,将0.1g的4-二甲氨基吡啶和1g的n,n'-二环己基碳二亚胺分别用10ml的n,n-二甲基甲酰胺溶解后逐滴加入到反应体系中,在机械搅拌下反应12h,将反应物抽滤,用无水乙醇洗涤3次,置于真空烘箱中60℃烘干,得到二氧化硅-聚丙烯酸中间体;胺基改性二氧化硅-聚丙烯酸中间体:称取10g的乙二胺,用100ml的n,n-二甲基甲酰胺将其溶解于500ml三口烧瓶中,置于冰浴中,在强搅拌下将二氧化硅-聚丙烯酸中间体的n,n-二甲基甲酰胺溶液逐滴加入到反应体系中,滴加完毕后继续反应24小时,将反应后的产物用抽滤瓶抽滤出来,用无水乙醇洗涤遍,最后置于60℃真空烘箱中干燥后得到最终产物。
42.实施例5
43.将40ml无水乙醇,4.46ml正硅酸乙酯及2g聚环氧乙烷加于三口瓶中,30℃下,在恒温磁力水浴锅中搅拌,至溶液均匀透明后,向反应釜中滴加去离子水,溶液立即变浑独,即发生溶胶凝胶反应,继续搅拌,3h后溶液呈半透明状态,取出静置,溶液分层,上层为乳白半透明溶液,下层析出物浓度较大呈微黄色;将下层微黄色析出物即聚环氧乙烷/二氧化硅混合液装入10ml的医用注射器中,医用注射器控制流速为0.5mm/s,进行电纺,纺丝条件为电压为12kv,接收距离为20cm,电纺得到聚乙烯醇/埃洛石纳米管改性复合纤维;取出纤维膜至铁盘中盛放,置于马弗炉内胆中,设定好升温程序后开始灼烧去除,升温程序为:以1.5℃/min的升温速率到200℃,保温2h,再以3.5℃/min的速率升高到600℃保温4h,随炉冷却。
44.称取3g二氧化硅纳米纤维膜,用100ml的n,n-二甲基甲酰胺溶解后倒入500ml三口烧瓶中超声分散30min,称取聚丙烯酸,用100ml的n,n-二甲基甲酰胺溶解后倒入三口烧瓶与二氧化硅纳米纤维混合,将混合溶液再超声分散30min后置于油浴中,将0.1g的4-二甲氨基吡啶和1g的n,n'-二环己基碳二亚胺分别用10ml的n,n-二甲基甲酰胺溶解后逐滴加入到反应体系中,在机械搅拌下反应12h,将反应物抽滤,用无水乙醇洗涤3次,置于真空烘箱中60℃烘干,得到二氧化硅-聚丙烯酸中间体;胺基改性二氧化硅-聚丙烯酸中间体:称取10g的聚丙胺,用100ml的n,n-二甲基甲酰胺将其溶解于500ml三口烧瓶中,置于冰浴中,在强搅拌下将二氧化硅-聚丙烯酸中间体的n,n-二甲基甲酰胺溶液逐滴加入到反应体系中,滴加完毕后继续反应24小时,将反应后的产物用抽滤瓶抽滤出来,用无水乙醇洗涤遍,最后置于60℃真空烘箱中干燥后得到最终产物。
45.对比例1
46.将40ml无水乙醇,4.46ml正硅酸乙酯及2g聚环氧乙烷加于三口瓶中,30-50℃下,在恒温磁力水浴锅中搅拌,至溶液均匀透明后,向反应釜中滴加去离子水,溶液立即变浑独,即发生溶胶凝胶反应,继续搅拌,3-5h后溶液呈半透明状态,取出静置,溶液分层,上层为乳白半透明溶液,下层析出物浓度较大呈微黄色;将下层微黄色析出物即聚环氧乙烷/二氧化硅混合液装入10ml的医用注射器中,医用注射器控制流速为0.5mm/s,进行电纺,纺丝条件为电压为12kv,接收距离为20cm,电纺得到聚乙烯醇/埃洛石纳米管改性复合纤维;取出纤维膜至铁盘中盛放,置于马弗炉内胆中,设定好升温程序后开始灼烧去除,升温程序为:以1.5℃/min的升温速率到200℃,保温2h,再以3.5℃/min的速率升高到600℃保温4h,随炉冷却。
47.对比例2
48.将40ml无水乙醇,4.46ml正硅酸乙酯及2g聚环氧乙烷加于三口瓶中,30℃下,在恒温磁力水浴锅中搅拌,至溶液均匀透明后,向反应釜中滴加去离子水,溶液立即变浑独,即发生溶胶凝胶反应,继续搅拌,3h后溶液呈半透明状态,取出静置,溶液分层,上层为乳白半透明溶液,下层析出物浓度较大呈微黄色;将下层微黄色析出物即聚环氧乙烷/二氧化硅混合液装入10ml的医用注射器中,医用注射器控制流速为0.5mm/s,进行电纺,纺丝条件为电压为12kv,接收距离为20cm,电纺得到聚乙烯醇/埃洛石纳米管改性复合纤维;取出纤维膜至铁盘中盛放,置于马弗炉内胆中,设定好升温程序后开始灼烧去除,升温程序为:以1.5℃/min的升温速率到200℃,保温2h,再以3.5℃/min的速率升高到600℃保温4h,随炉冷却。
49.称取3g二氧化硅纳米纤维膜,用100ml的n,n-二甲基甲酰胺溶解后倒入500ml三口烧瓶中超声分散30min,称取聚丙烯酸,用100ml的n,n-二甲基甲酰胺溶解后倒入三口烧瓶与二氧化硅纳米纤维混合,将混合溶液再超声分散30min后置于油浴中,将0.1g的4-二甲氨基吡啶和1g的n,n'-二环己基碳二亚胺分别用10ml的n,n-二甲基甲酰胺溶解后逐滴加入到反应体系中,在机械搅拌下反应12h,将反应物抽滤,用无水乙醇洗涤3次,置于真空烘箱中60℃烘干,得到二氧化硅-聚丙烯酸中间体。
50.图1、图2、图3分别为本发明制备的实施例1-3中二氧化硅纳米纤维的扫描图,从图中可以看出,制备的二氧化硅纳米纤维呈现一种多孔、中空的纳米纤维。
51.二氧化碳吸附性能测试结果
52.表1
[0053] 孔隙率(cm3/g)比表面积(m2/g)实施例10.62
±
0.0831.6
±
7.6实施例20.63
±
0.1132.3
±
4.8实施例30.62
±
0.0932.5
±
6.3实施例40.75
±
0.1335.0
±
3.5实施例50.81
±
0.0631.8
±
7.6对比例11.05
±
0.07256.3
±
11.2对比例21.26
±
0.11335.8
±
9.3
[0054]
表2
[0055]
[0056][0057]
表1为本发明制备的实施例1-5、对比例1和2的孔隙率和比表面积,从表中可以看出,对比例2的孔隙率和比表面积均好于实施例1-5。
[0058]
图4为本发明制备的对比例1(a)和实施例1(b)在40℃条件下的二氧化碳吸附曲线。图5为本发明制备的实施例1和对比例1和2制得的二氧化碳吸附曲线,从图中可以看出,实施例1的吸附容量要远远高于对比例和对比例2的吸附容量。再结合表1中的数据,分析原因可能是聚丙烯酸的接枝影响到了纳米纤维的孔隙率,但以聚丙烯酸分子链中大量的羧基为活性点,取代二氧化硅表面有限的羟基基团与端胺基的分子反应,从而可以大量增加胺基的引入两,进一步的增大吸附剂的吸附容量。
[0059]
图6为本发明制备的实施例1进行吸附循环测试制得的吸附容量图。从图中可以看出吸附剂的二氧化碳吸附量虽然有少许的下降,但是仍保持在较高的水平,说明此种吸附剂可再生性比较好。
[0060]
图7为本发明制备的实施例1对二氧化碳和氮气的吸附平衡曲线。从图中可以看出,当实施例1制备的吸附剂用于吸附二氧化碳时,30分钟后就达到吸附平衡,与传统的醇胺溶液相比,实施例1的吸附剂吸附速率较慢,原因可能是醇胺溶液中的胺基空间均匀分布,能和二氧化碳进行完全接触,而本发明制备的吸附剂,胺基负载在固体基上,空间分布不均匀,刚开始只能与少量的二氧化碳接触。但是本发明制备的吸附剂对于氮气的吸附量特别的少,几乎没有,因此本发明制备的吸附剂的气体选择吸附效果比较好。
[0061]
最后应说明的是:以上所述实施例仅表达了本发明的具体实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1